Tutorial¶
This tutorial is intended as an introduction to working with MetagenomeDB.
Before we start, make sure that you have the MetagenomeDB toolkit installed (see Installation). In the Python shell, the following should run without raising an exception:
>>> import MetagenomeDB
For practical purpose, in all examples of this tutorial we will use an alias for the library, mdb:
>>> import MetagenomeDB as mdb
This tutorial also assumes that a MongoDB server is running and the connection information (server host, port, name of the database, etc.) are stored in the file ~/.MetagenomeDB. Assuming you have downloaded and installed MongoDB, you can start it like so:
$ mongod
Example¶
MetagenomeDB allows scientists to store large amount of sequences, such as those generated by genomics and metagenomics projects. Sequences can be grouped into collections (see Introduction). Collections can be connected to each others (see Relationships). Finally, both sequences and collections can be annotated with arbitrary information (see Annotations).
Let’s start with an example. Imagine you have collected a biological sample from an environment (soil, water, etc.), extracted some sub-samples using various purification techniques, and performed a metagenomic analysis on these sub-samples (i.e., generated reads and assembled them into contigs). MetagenomeDB will help you organizing this wealth of information by representing it according to the following structure:
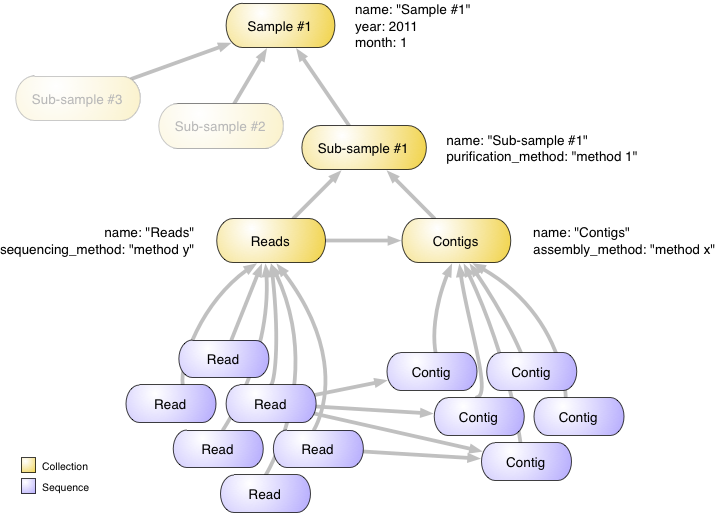
At the top of this structure we have an object (a Collection) that represents your biological sample. We annotate it with a name (‘Sample #1’), and the month and year this sample was collected (month and year properties). You could easily add any other annotation; e.g., who collected the sample, using which sampling technique (see Annotations).
Right under this structure we have various sub-samples, also represented as collections. For display purpose, we will only focus on one of these. This sub-sample receives a name (‘Sub-sample #1’) and a description of the purification method used (purification_method property).
This sub-sample has been sequenced, which means a set of reads has been obtained. Those reads have further been assembled into contigs. Both reads and contigs are represented by numerous Sequence objects. To better organize these two sets of sequences we create two collections, named ‘Reads’ and ‘Contigs’. The ‘Reads’ collection contains information about how the sequencing was made, while the ‘Contigs’ collection contains information about how the assembly was made.
Arrows in this figure represent relationships (see Relationships) between objects. Hence, reads and contigs are related to their respective collections. Reads are also related to contigs to express the fact that some of the reads were used to assemble some of the contigs. Finally, collections are related to each other to represent how they are hierarchically organized. Think of an arrow as a ‘part of’ relationship: a read is part of the Reads collection, which itself is part of a sub-sample (and also part of the Contigs collection), which itself is part of a sample.
Note
The organization described here is completely arbitrary; MetagenomeDB can manage any number of collections and sequences you need, and any relationships between them. However, this specific organization proved the most convenient when used by biologists, and you may want to use it as a template for your own data.
Let see how to create such a structure and import your data.
Creating and linking collections¶
The first step in creating the structure described above is to define a set of collections and their relationships. This can be done either programmatically using the API (see API Documentation), or through command-line tools (see Tools Documentation).
Using command-line tools¶
The first option to create your collections is to use the mdb-add command-line tool (see mdb-add: Adding objects) together with a text file (either a CSV file generated by a spreadsheet software, or a JSON file; see the tool documentation) that describes the objects and their relationships.
A CSV (comma separated values) file can be created by any spreadsheet software that supports this format, such as Microsoft Excel, OpenOffice or Numbers.
In this file each line will describe a single object, or a relationship between two objects. Each cell of this line will contain an annotation for this object or relationship.
For example, creating the collection ‘Sample #1’ requires the following following table:
| A | B | C | D |
|---|---|---|---|
| _type=collection | name=Sample #1 | year=2011^integer | month=1^integer |
Note
The header A B C... is only for display purpose; you do not have to add this line to your file.
Please note that:
- the order of the cells in each line is not important
- each cell follows the same syntax: [property name] = [value of this property] [^optional modifier] (spaces around the equal sign or after the value are ignored). The modifier is needed when the value of your property is not a string (as for the name property). Hence, the year and month properties are both integers. For a more complete reference, see mdb-add: Adding objects.
- one of the cell has to declare the special property _type to tell mdb-add the type of object to create. Only three values are accepted: collection, sequence and relationship (case-insensitive). All properties with a starting underscore are special properties that are used by the MetagenomeDB API; you cannot use those properties to annotate your objects, or modify them.
- one of the cell has to declare the property name. All collections and sequences in MetagenomeDB have at least a name; sequences also require a sequence property (see Collection and Sequence).
Creating the sub-sample ‘Sub-sample #1’ just means adding another line:
| A | B | C | D |
|---|---|---|---|
| _type=collection | name=Sample #1 | year=2011^integer | month=1^integer |
| _type=collection | name=Sub-sample #1 | purification_method=method #1 |
Finally, to connect the sub-sample to its sample, you need to declare a relationship:
| A | B | C | D | E |
|---|---|---|---|---|
| _type=collection | name=Sample #1 | year=2011^integer | month=1^integer | |
| _type=collection | name=Sub-sample #1 | purification_method=method #1 | ||
| _type=relationship | _source._type=collection | _source.name=Sub-sample #1 | _target._type=collection | _target.name=Sample #1 |
Relationships have special properties:
- two special properties _source and _target must be provided to describe the source and the target of this relationship, respectively. Remember that relationships are always directed in MetagenomeDB (see Relationships). In our case, Sub-sample #1 is related to (is part of) Sample #1, but not the other way around.
- _source and _target must provide two sub-properties, name and _type. name is the name of the object you want to connect, while _type is its type. In our example, we want to connect a collection to another. Connecting a sequence to a collection (or a sequence to another sequence) would require a third sub-property, _collection, to indicate which collection this sequence belong to. However adding sequences to a collection is typically done using the mdb-import-sequences command-line tool, described below.
That’s it! This is the only thing you need to know to describe your collections and their relationships. Here is the final table, including the ‘Reads’ and ‘Contigs’ collections. This table is also available as a download: table.csv.
| A | B | C | D | E |
|---|---|---|---|---|
| _type=collection | name=Sample #1 | year=2011^integer | month=1^integer | |
| _type=collection | name=Sub-sample #1 | purification_method=method #1 | ||
| _type=relationship | _source._type=collection | _source.name=Sub-sample #1 | _target._type=collection | _target.name=Sample #1 |
| _type=collection | name=Reads | sequencing_method=method y | ||
| _type=collection | name=Contigs | assembly_method=method x | ||
| _type=relationship | _source._type=collection | _source.name=Reads | _target._type=collection | _target.name=Sub-sample #1 |
| _type=relationship | _source._type=collection | _source.name=Contigs | _target._type=collection | _target.name=Sub-sample #1 |
| _type=relationship | _source._type=collection | _source.name=Reads | _target._type=collection | _target.name=Contigs |
From there you can call mdb-add with the name of your CSV file as an argument
$ mdb-add -i table.csv
The output should be:
4 collections added
4 relationships added
Your objects are now in the database. Later you will want to look at the documentation for mdb-add (mdb-add: Adding objects) to know how to deal with special cases, such as duplicate objects, and to learn more about the syntax for property values.
Using the API¶
The same structure can be created using the MetagenomeDB API; i.e., by writing a Python script. Here is the equivalent of the above structure declaration using the API:
import MetagenomeDB as mdb
# first, we create a new Collection object for the sample. The Collection()
# class takes as an argument a dictionary with all properties you want to
# annotate this collection with.
sample = mdb.Collection({"name": "Sample #1", "year": 2011, "month": 1})
# once created, you need to commit this collection to the database.
sample.commit()
# similarly, we create a Collection object for the sub-sample
subsample = mdb.Collection({"name": "Sub-sample #1", "purification_method": "method #1"})
# we then link this sub-sample to the sample
subsample.add_to_collection(sample)
# then we commit it
subsample.commit()
# same thing for the 'Reads' and 'Contigs' collections:
reads = mdb.Collection({"name": "Reads", "sequencing_method": "method y"})
contigs = mdb.Collection({"name": "Contigs", "assembly_method": "method x"})
contigs.add_to_collection(subsample)
contigs.commit()
# please note here that 'contigs' needs to be committed before
# we relate 'reads' to it:
reads.add_to_collection(subsample)
reads.add_to_collection(contigs)
reads.commit()
For a more complete documentation of the API, please refer to the API Documentation.
Importing sequences¶
Once your collections have been created you are ready to import sequences; in our examples, the sets of reads and contigs generated by a metagenomic analysis.
Note
Sequences do not have to be related to a collection in MetagenomeDB. However this is extremely convenient to describe them as a set (e.g., the set of all reads generated by a sequencing run). Also, all command-line tools dealing with sequences will expect them to be part of a collection.
You could use the same approach as above; i.e., creating a CSV file with a sequence description per line (with properties such as the nucleotide sequence, the sequence name, etc.) and a relationship between this sequence and the collection (either ‘Reads’ or ‘Contigs’), then importing this file by calling mdb-add. However, a dedicated tool is provided to import sequences from a variety of file formats: mdb-import-sequences (see EMPTY).
mdb-import-sequences can read nucleotide and protein sequences from FASTA files as well as all formats supported by the Biopython library, such as GenBank or FASTQ (for a complete list, please refer to the Biopython documentation).
Importing a set of reads from a file reads.fasta into the ‘Reads’ collection is achieved by typing the following on the command-line:
$ mdb-import-sequences -i reads.fasta -C Reads
Similarly for contigs in a contigs.fasta file:
$ mdb-import-sequences -i contigs.fasta -C Contigs
mdb-import-sequences comes with extra features, such as annotating all sequences from the same file with a same property and value. This can be useful, for example, when you want to add a property class with value ‘read’ for all reads, or ‘contig’ for all contigs. The syntax becomes:
$ mdb-import-sequences -i reads.fasta -C Reads -c class read
$ mdb-import-sequences -i contigs.fasta -C Contigs -c class contig
For a complete documentation of mdb-import-sequences, see EMPTY.
Connecting sequences¶
The reads and contigs you just imported are not disconnected. In fact, for each contig there is a set of reads that has been used for its assembly. Again, you could import this information as a set of relationships defined in a CSV file. But another tool, mdb-import-ACE-alignments (see EMPTY) is available to import reads-to-contig mapping in the ACE format, often generated by assembly programs.
Hence, to import an assembly file mapping.ace you need to provide the name of the collection that contains your reads and the one containing your contigs:
$ mdb-import-ACE-alignments -i mapping.ace -R Reads -C Contigs
mdb-import-ACE-alignments comes with several features, such as ignoring missing reads or contigs (i.e., reads or contigs defined in the ACE file but not present in the database) or converting on-the-fly the read or contig identifiers. For a complete documentation, see EMPTY.
Modifying existing objects¶
We saw earlier how to define collections and/or sequences using a CSV file and the mdb-add tool, or by using the API. However, what about modifying objects that have been already created? You can add, modify (i.e., replace, append to) or remove any property of an object by using either the command line or the API.
Using the command line¶
The mdb-annotate tool (Annotating objects) is available to modify existing objects, using the same approach as mdb-add; i.e., by creating a CSV or JSON file with instructions for these objects.
The syntax of this file is the same as for mdb-add. The only difference is the availability of operators. Hence, the following CSV file will replace the property year of the collection ‘Sample #1’ by the value 2010:
| A | B | C |
|---|---|---|
| _type=collection | name=Sample #1 | year=2010^integer |
What next?¶
Done. All your data are now in the database. At this stage you can query the database (see Queries) and/or export some of its content (see Tools Documentation). In the Examples section you will find some examples of such activities, such as managing sequence alignments (as produced by BLAST or FASTA) or producing Cytoscape networks out of your data.