
Project Summary
Description: This is a real world application of Leaf. It is the protocol for a CNV study in a rare disease. Details
can't be disclosed since the study is still unpublished at the moment of publishing its protocol as a Leaf
example. For the same reason, links to output files are intentionally not active!.
Number of nodes: 40
Number of [F]-nodes: 21
Total number of output files: 122
Total size of output files: 3.48G
Total CPU time required: 01:03:31.05
Number of nodes: 40
Number of [F]-nodes: 21
Total number of output files: 122
Total size of output files: 3.48G
Total CPU time required: 01:03:31.05
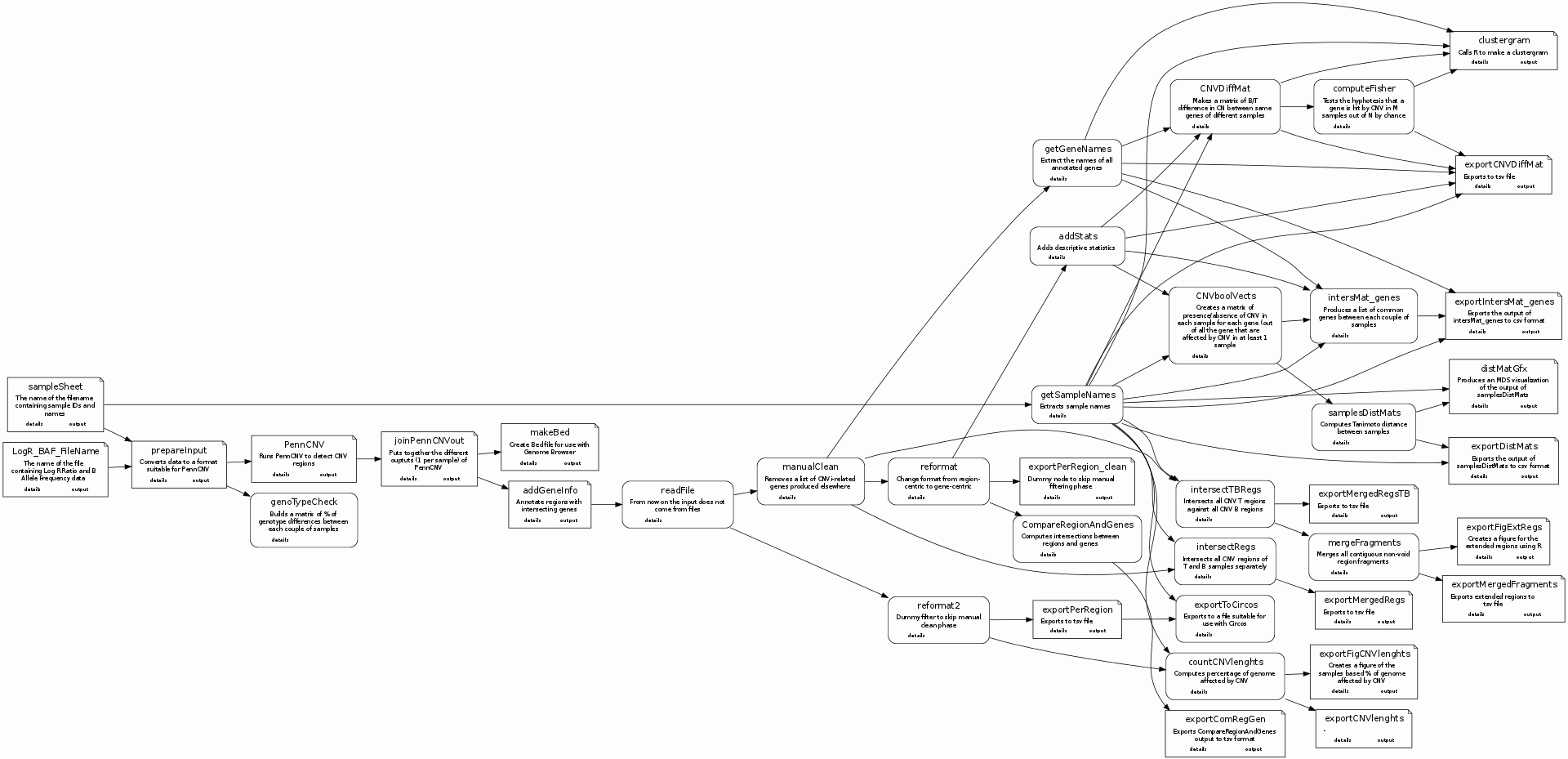
Protocol map
Nodes details
exportComRegGen
Exports CompareRegionAndGenes output to tsv format
Output file: CompareRegGenes.txt
Last build was: Sun Jan 8 03:47:10 2012.
Required CPU time: 00:00:0.08.
def exportComRegGen(data):
"""Exports CompareRegionAndGenes output to tsv format"""
f = open('CompareRegGenes.txt','w')
f.write('Gene\tSample\ts/t\tReg. start\tReg. end\tReg. size\tTanscript. start\t Transcript. end\tTranscript. size\tCode start\t Code end\tCode size\tInters. to Transcr. %\t Inters. to Code %')
for line in data:
if line[0]!='NOT_FOUND':
for item in line:
if type(item)==float:
f.write(str(item)+'\t')
f.write('\n')
return 'CompareRegGenes.txt'
exportMergedRegs
Exports to tsv file
Output file: mergedRegsTB.tsv
Last build was: Sun Jan 8 03:47:16 2012.
Required CPU time: 00:00:0.13.
def exportMergedRegs(mergedRegs):
"""Exports to tsv file"""
f = open('mergedRegsTB.tsv', 'w')
f.write('B/T\tChr\tReg. Start\tReg. End\tNum Samples\tCN\t"Genes"\n')
for key in sorted(mergedRegs.keys()):
for i in range(0, len(mergedRegs[key][0])):
try:
f.write(('B' if key[0] == 's' else 'T') +'\t'+
key[1]+'\t'+
str(mergedRegs[key][0][i]).strip('[]').replace(', ','\t')+'\t'+
str(mergedRegs[key][1][i])+'\t'+
str(mergedRegs[key][3][i]).
replace('[','').
replace(']','').
replace("'", '')+'\t'+
str(mergedRegs[key][2][i]).replace('set','').
replace(')','').
replace('(','').
replace('[','').
replace(']','').
replace("'", '')+'\n'
)
except: import pdb; pdb.set_trace()
return 'mergedRegsTB.tsv'
exportIntersMat_genes
Exports the output of intersMat_genes to csv format
Output file: verbose_inters_mat.txt
Last build was: Sun Jan 8 03:47:20 2012.
Required CPU time: 00:00:0.08.
def exportIntersMat_genes(genes, samples, inters_names):
"""Exports the output of intersMat_genes to csv format"""
f = open('verbose_inters_mat.txt', 'w')
f.write(' ,'+','.join(map(lambda x:x[0]+x[1], samples)) + '\n')
for idx1 in range(0,len(inters_names)):
f.write(samples[idx1][0]+samples[idx1][1]+',')
for idx2 in range(0,len(inters_names)):
f.write(str(list(inters_names[idx1][idx2])).strip('[]').replace('\'','').replace(',','') + ',')
f.write('\n')
return 'verbose_inters_mat.txt'
joinPennCNVout
Puts together the different ouptuts (1 per sample) of PennCNV
Output file: pglfeb.csv_allNO14.out
Last build was: Sun Jan 8 03:45:59 2012.
Required CPU time: 00:00:0.11.
def joinPennCNVout(inputfiles):
"""Puts together the different ouptuts (1 per sample) of PennCNV"""
#log_ofile = open(fname + '_all.log', 'a')
if os.path.exists(fname + '_allNO14.out'):
os.remove(fname+'_allNO14.out')
out_ofile = open(fname + '_allNO14.out', 'a')
for ifile in inputfiles:
#log_ifname = ifile + '.log'
#log_ofile.write(open(log_ifname, 'r').read())
#if not '14B' in ifile and not '5B' in ifile and not '34' in ifile and not '33' in ifile:
#if not '14' in ifile and not '21' in ifile and not '14B' in ifile and not '5B' in ifile:
out_ofile.write(open(ifile, 'r').read())
out_ofile.close()
return fname + '_allNO14.out'
exportPerRegion
Exports to tsv file
Output file: perRegion.csv
Last build was: Sun Jan 8 03:51:58 2012.
Required CPU time: 00:00:0.26.
def exportPerRegion(data):
"""Exports to tsv file"""
f = open('perRegion.csv','w')
sdict = idToNameDict()
f.write(data[0].replace('\t',',')+'\n')
for line in data[1:]:
l = line.replace(',','.').split()
l[6] = sdict[l[6]]
l = str(l).replace('\'', '').strip('[]')
f.write(l+'\n')
return 'perRegion.csv'
addGeneInfo
Annotate regions with intersecting genes
Output file: pglfeb.csv_allNO14.out.genes
Last build was: Sun Jan 8 03:46:51 2012.
Required CPU time: 00:00:51.95.
def addGeneInfo(fname):
"""Annotate regions with intersecting genes"""
#print 'SKIPPING'
#return fname + '.genes'
os.system('perl ' + penncnv_path + '/scan_region.pl "' + fname + '" -refgene ' + annot_path + '/refGene.txt -reflink ' + annot_path + '/refLink.txt > "' + fname + '.genes1"')
os.system('perl ' + penncnv_path + '/scan_region.pl "' + fname + '.genes1" -refgene ' + annot_path + '/refGene.txt -reflink ' + annot_path + '/refLink.txt -expandleft 5m > "' + fname + '.genes2"')
os.system('perl ' + penncnv_path + '/scan_region.pl "' + fname + '.genes2" -refgene ' + annot_path + '/refGene.txt -reflink ' + annot_path + '/refLink.txt -expandright 5m > "' + fname + '.genes"')
os.system('rm "' + fname + '".genes1')
os.system('rm "' + fname + '".genes2')
#os.system('perl ' + penncnv_path + '/detect_cnv.pl -cctest -pfb ' + penncnv_path + '/lib/ho1v1.hg18.pfb -phenofile phenotype -cnv "' + fname + '" > "' + fname + '".CC')
return fname + '.genes'
sampleSheet
The name of the filename containing sample IDs and names
Output file: sample_sheet.tsv
Last build was: Sun Jan 8 03:37:36 2012.
Required CPU time: 00:00:0.03.
def sampleSheet():
"""The name of the filename containing sample IDs and names"""
return('sample_sheet.tsv')
readFile
From now on the input does not come from files
Last build was: Sun Jan 8 03:46:51 2012.
Required CPU time: 00:00:0.06.
def readFile(fname):
"""From now on the input does not come from files"""
return open(fname).readlines()
distMatGfx
Produces an MDS visualization of the output of samplesDistMats
Output files: t_tani_distrib.pdf , t_tani_mds.pdf .
Last build was: Sun Jan 8 03:51:56 2012.
Required CPU time: 00:00:0.25.
def distMatGfx(samples, mats):
"""Produces an MDS visualization of the output of samplesDistMats"""
tani = mats[2]
test_couples=[['13t','14t'], ['33t1','33t2'], ['34t','34tL'], ['5t', '5Bt']]
#s = getSampleNames()
s = samples
tani_t = list()
tani_t_mat = list()
tids = list()
print 'Adding background distances :'
for idx1, s1 in enumerate(s):
matrow = list()
if 't' in s1:
tids.append(s1)
for idx2, s2 in enumerate(s):
if 't' in s2:
matrow.append(tani[idx1,idx2])
if s1 != s2:
tani_t.append(tani[idx1,idx2])
#print s[idx1] + '-' + s[idx2]+',',
tani_t_mat.append(matrow)
#print
#hist(tani_t,30)
test_dists=list()
sdict = idToNameDict()
for idx, item in enumerate(test_couples):
if item[0] not in s or item[1] not in s:
continue
s1 = s.index(item[0])
s2 = s.index(item[1])
d = tani[s1,s2]
test_dists.append(d)
#annotate('('+sdict[item[0]]+','+sdict[item[1]]+')', [d,10], [d,10*idx+20])
print d
mds=r.cmdscale(array(tani_t_mat))
#savefig('t_tani_distrib.pdf')
#figure()
#plot(mds[:,0], mds[:,1], '.')
for idx, item in enumerate(tids):
pass
#annotate(sdict[item], [mds[idx,0], mds[idx,1]])
#savefig('t_tani_mds.pdf')
return ['t_tani_distrib.pdf', 't_tani_mds.pdf']
intersectTBRegs
Intersects all CNV T regions against all CNV B regions
Last build was: Sun Jan 8 19:44:57 2012.
Required CPU time: 00:00:7.36.
def intersectTBRegs(getSampleNames, manualClean):
"""Intersects all CNV T regions against all CNV B regions"""
f,h = PennCNVParser()
manualClean = [f.split(l) for l in manualClean]
num = dict()
num['s'] = sum(['s' in s for s in getSampleNames])
num['t'] = sum(['t' in s for s in getSampleNames])
regsets = dict()
chrs = set([l[1] for l in manualClean])
for samp in getSampleNames:
for chrom in chrs:
sub = [l for l in manualClean if l[7]==samp and l[1] == chrom]
st = 'B' if 's' in samp else 'T'
regset = [[int(l[2]),int(l[3]), l[10], l[6], st] for l in sub]
if not chrom in regsets.keys():
regsets[chrom] = list()
if not sub == []:
regsets[chrom].append(regset)
a = dict()
for chrom in chrs:
# if chrom == '13':
# import pdb; pdb.set_trace()
a[chrom]=intersRegs([sorted(s) for s in regsets[chrom]])
return a
exportToCircos
Exports to a file suitable for use with Circos
Last build was: Sun Jan 8 03:51:59 2012.
Required CPU time: 00:00:0.02.
def exportToCircos(samples, x):
"""Exports to a file suitable for use with Circos"""
return "tobefixed!"
x=[l.split('\t') for l in x][1:]
x=[[l[0],l[1],l[2],l[5],l[6]] for l in x]
fnames = list()
for sample in samples:
fname = 'circosInput_' + sample + '.txt'
fnames.append(fname)
f = open(fname, 'w')
subx = [l[0:4] for l in x if l[4]==sample]
f.writelines("hs%s\n" % str(item).strip('[]').replace('\'','').replace(',','') for item in subx)
return(fnames)
intersectRegs
Intersects all CNV regions of T and B samples separately
Last build was: Sun Jan 8 03:47:15 2012.
Required CPU time: 00:00:5.19.
def intersectRegs(getSampleNames, manualClean):
"""Intersects all CNV regions of T and B samples separately"""
f,h = PennCNVParser()
manualClean = [f.split(l) for l in manualClean]
num = dict()
num['s'] = sum(['s' in s for s in getSampleNames])
num['t'] = sum(['t' in s for s in getSampleNames])
regsets = dict()
chrs = set([l[1] for l in manualClean])
for samp in getSampleNames:
for chrom in chrs:
sub = [l for l in manualClean if l[7]==samp and l[1] == chrom]
regset = [[int(l[2]),int(l[3]), l[10], l[6]] for l in sub]
if 's' in samp:
if not ('s',chrom) in regsets.keys():
regsets['s', chrom] = list()
regsets['s',chrom].append(regset)
else:
if not ('t',chrom) in regsets.keys():
regsets['t', chrom] = list()
regsets['t',chrom].append(regset)
a = dict()
for chrom in chrs:
a['s',chrom]=intersRegs([sorted(s) for s in regsets['s',chrom]])
a['t',chrom]=intersRegs([sorted(s) for s in regsets['t',chrom]])
return a
reformat2
Dummy filter to skip manual clean phase
Last build was: Sun Jan 8 03:51:58 2012.
Required CPU time: 00:00:1.83.
def reformat2(ifile):
'Dummy filter to skip manual clean phase'
f, header = PennCNVParser()
ofile = list()
ofile.append('\t'.join(header))
p = getPairings()
for line in ifile:
newline = (f.split(line)[1:-1])
try:
t=newline.pop(10)
except IndexError:
print line
if newline[6] in [x[0] for x in p]:
newline.insert(7,'s')
else:
newline.insert(7,'t')
genes = newline[10].split(',')
for gene in genes:
newline[10]=gene
ofile.append('\t'.join(newline))
return ofile
genoTypeCheck
Builds a matrix of % of genotype differences between each couple of samples
Last build was: Sun Jan 8 19:42:16 2012.
Required CPU time: 00:02:57.52.
def genoTypeCheck(fnames):
"""Builds a matrix of % of genotype differences between each couple of samples"""
data=[]
for ifile in fnames:
curgtype = list()
cursamp = open(ifile).readlines()
for line in cursamp:
line = line.split('\t')
curgtype.append(line[3])
data.append(curgtype)
data=array(data)
differences=list()
f = open('gtypecheck_small.txt', 'w')
sdict = idToNameDict()
for sample1 in data:
templist = list()
for sample2 in data:
templist.append(float(sum(array(sample1)!=array(sample2))) / len(sample1) * 100)
try:
print sdict[sample1[0].replace('.GType','')], 'vs', sdict[sample2[0].replace('.GType','')],':','\t', templist[-1], '\t'
#print templist[-1]
if templist[-1] < 20 and sdict[sample1[0].replace('.GType','')] != sdict[sample2[0].replace('.GType','')]:
f.write(sdict[sample1[0].replace('.GType','')] + ' vs ' + sdict[sample2[0].replace('.GType','')] + '\t' + str(templist[-1]) + '\n')
differences.append(templist)
f = open('gtypecheck.txt', 'w')
for idx, item in enumerate(differences):
f.write(sdict[data[idx][0].replace('.GType','')]+' ')
f.write('\n')
for idx, item in enumerate(differences):
f.write(' ' + sdict[data[idx][0].replace('.GType','')])
f.writelines(str(array(item)).strip('[]').replace('\n','')+'\n')
except:
print 'Skipping couple: ' + sample1[0] +', '+sample2[0]
return differences
getGeneNames
Extract the names of all annotated genes
Last build was: Sun Jan 8 03:47:16 2012.
Required CPU time: 00:00:0.05.
def getGeneNames(f):
"""Extract the names of all annotated genes"""
data=list()
for line in f:
nline=line[0:-1]
t = nline.split()
data.extend(t[7].split(','))
data=array(data)
return unique(data)
countCNVlenghts
Computes percentage of genome affected by CNV
Last build was: Sun Jan 8 19:43:38 2012.
Required CPU time: 00:00:0.36.
def countCNVlenghts(getSampleNames, reformat):
"""Computes percentage of genome affected by CNV"""
di = idToNameDict()
y=[l.split('\t') for l in reformat]
glength=float(2867188341)
res = []
for s in getSampleNames:
try:
sub=[l for l in y if l[6]==s]
lens = [int(l[2])-int(l[1]) for l in sub]
res.append([di[s] , str(sum(lens)) , str(100*(sum(lens) / glength))])
except:
import pdb; pdb.set_trace()
for st in ['s', 't']:
sub = [l for l in y if l[7]==st]
lens = [int(l[2])-int(l[1]) for l in sub]
if st=='s':
label = 'Blood'
else:
label = 'Tumor'
print label + ' average: ' + str(sum(lens) /
(len(getSampleNames)/float(2))) +'\t' + str(100 * (sum(lens)/ (len(getSampleNames)/float(2)) / glength) )
return res
addStats
Adds descriptive statistics
Last build was: Sun Jan 8 03:47:17 2012.
Required CPU time: 00:00:0.99.
def addStats(fname):
"""Adds descriptive statistics"""
data=list()
f = fname
for line in f:
line=line[0:-1]
data.append(line.split('\t'))
data=array(data)
genes = unique(data[:,10])
p = getPairings()
s=list()
for samp in p[0]:
for raw in data:
if raw[6] == samp:
s.append(raw)
s=array(s)
t=list()
for samp in p[1]:
for raw in data:
if raw[6] == samp:
t.append(raw)
t=array(t)
gene_count = dict()
for gene in genes:
if gene == 'NOT_FOUND':
gene_count[gene]=(0,0,0,0)
else:
s_where = s[nonzero(s[:,10]==gene)]
t_where = t[nonzero(t[:,10]==gene)]
s_count = sum(s[:,10]==gene)
t_count = sum(t[:,10]==gene)
s_diffcount = len(unique(s_where[:,6]))
t_diffcount = len(unique(t_where[:,6]))
gene_count[gene]=(s_count, t_count, s_diffcount, t_diffcount)
of=list()
for idx, line in enumerate(data):
if idx==0:
of.append('\t'.join(line) + '\ts hits \tt hits\thits diff.\ts un. hits\tt un. hits\t un. hits diff.')
else:
currinfo = gene_count[line[10]]
of.append('\t'.join(line)+\
'\t'+str(currinfo[0])+\
'\t'+str(currinfo[1])+\
'\t'+str(abs(currinfo[0]-currinfo[1]))+\
'\t'+str(currinfo[2])+\
'\t'+str(currinfo[3])+\
'\t'+str(abs(currinfo[2]-currinfo[3])))
#'\t'+str(pvalue(currinfo[2], 8-currinfo[2], currinfo[3], 8-currinfo[3]).two_tail)+'\n')
return of
exportCNVlenghts
[documentation not available]
Output file: cnv_lenghts.tsv
Last build was: Sun Jan 8 19:43:38 2012.
Required CPU time: 00:00:0.02.
def exportCNVlenghts(countCNVl):
f = open('cnv_lenghts.tsv', 'w')
for line in countCNVl:
f.writelines(str(line).strip('[]').replace(',','\t').replace("'",'')+'\n')
return 'cnv_lenghts.tsv'
exportDistMats
Exports the output of samplesDistMats to csv format
Output files: inters_mat.txt , notIntersMat.txt , tanimotoMat.txt .
Last build was: Sun Jan 8 19:43:38 2012.
Required CPU time: 00:00:0.03.
def exportDistMats(samples, mats):
"""Exports the output of samplesDistMats to csv format"""
intMat=mats[0]
notIntMat=mats[1]
tanimotoMat=mats[2]
f_intMat = open('intersMat.txt','w')
f_notIntMat = open('notIntersMat.txt','w')
f_tanimotoMat = open('tanimotoMat.txt','w')
f_intMat.write(' ,'+','.join(samples) + '\n')
f_notIntMat.write(' ,'+','.join(samples) + '\n')
f_tanimotoMat.write(' ,'+','.join(samples) + '\n')
for idx, item in enumerate(intMat):
f_intMat.write(samples[idx]+',')
f_intMat.write(str(list(intMat[idx])).strip('[]').replace('.0','').replace(' ','')+'\n')
f_notIntMat.write(samples[idx]+',')
f_notIntMat.write(str(list(notIntMat[idx])).strip('[]').replace('.0','').replace(' ','')+'\n')
f_tanimotoMat.write(samples[idx]+',')
f_tanimotoMat.write(str(list(tanimotoMat[idx])).strip('[]').replace(' ','')+'\n')
return ['inters_mat.txt', 'notIntersMat.txt', 'tanimotoMat.txt']
intersMat_genes
Produces a list of common genes between each couple of samples
Last build was: Sun Jan 8 03:47:19 2012.
Required CPU time: 00:00:0.38.
def intersMat_genes(dataset, fname, genes, samples):
"""Produces a list of common genes between each couple of samples"""
inters_names = list()
for idx1, geneline1 in enumerate(dataset):
inters_names_t = list()
for idx2, geneline2 in enumerate(dataset):
inters_names_t.append(genes[nonzero(logical_and(geneline1, geneline2))])
inters_names.append(inters_names_t)
return inters_names
reformat
Change format from region-centric to gene-centric
Last build was: Sun Jan 8 03:46:56 2012.
Required CPU time: 00:00:0.69.
def reformat(ifile):
"""Change format from region-centric to gene-centric"""
f, header = PennCNVParser()
ofile = list()
ofile.append('\t'.join(header))
p = getPairings()
for line in ifile:
newline = (f.split(line)[1:-1])
try:
t=newline.pop(10)
except IndexError:
print line
if newline[6] in [x[0] for x in p]:
newline.insert(7,'s')
else:
newline.insert(7,'t')
genes = newline[10].split(',')
for gene in genes:
newline[10]=gene
ofile.append('\t'.join(newline))
return ofile
CNVboolVects
Creates a matrix of presence/absence of CNV in each sample for each gene (out of all the gene that are affected by CNV in at least 1 sample
Last build was: Sun Jan 8 03:47:18 2012.
Required CPU time: 00:00:0.35.
def CNVboolVects(raw, allsamples):
"""Creates a matrix of presence/absence of CNV in each sample for each gene (out of all the gene that are affected by CNV in at least 1 sample"""
data=list()
f=raw
for line in f:
line=line[0:-1]
data.append(line.split('\t'))
data=array(data)
genes = unique(data[1:-1,10])
hashd = dict( zip( genes, range( 0,len(genes) ) ) )
#samples = getSampleNames()
samples = allsamples
s=data
vectors = np.zeros((len(samples),len(genes)))
for s_idx, sample in enumerate(samples):
sample_genes = s[s[:,6]==sample][:,10]
for gene in sample_genes:
vectors[s_idx][hashd[gene]]=1
return vectors
LogR_BAF_FileName
The name of the file containing Log R Ratio and B Allele Frequency data
Output file: pglfeb.csv
Last build was: Sun Jan 8 03:37:36 2012.
Required CPU time: 00:00:0.03.
def LogR_BAF_FileName():
"""The name of the file containing Log R Ratio and B Allele Frequency data"""
return(fname)
clustergram
Calls R to make a clustergram
Output files: clustergram_001_108.png , clustergram_005_248.png , clustergram_05_1606.png , heatmap_001_108.png , heatmap_005_248.png , heatmap_05_1606.png .
Last build was: Sun Jan 8 19:44:49 2012.
Required CPU time: 00:00:57.72.
def clustergram(diffmat, fisher, genes, samples):
"""Calls R to make a clustergram"""
#samplesNoLetter = [item.replace('t','').replace('s','') for item in samples]
#samples = []
#[samples.append(i) for i in samplesNoLetter if not i in samples]
p=getPairings()
sdict = idToNameDict()
s2 = list()
for i in range(0,len(p)):
s2.append(sdict[p[i][0]]+' vs '+sdict[p[i][1]])
samples = s2
d=diffmat[0]
gdict = geneChrDict()
#import pdb; pdb.set_trace()
thresholds = [0.01, 0.05, 0.5]
cfnames = list()
hfnames = list()
for t in thresholds:
ok_genes=list()
ok_gnames=list()
for idx, gene_row in enumerate(d):
if fisher[1][idx] < t:
if str.lower(genes[idx]) != 'loc100130872-spon2' and str.lower(genes[idx]) != 'loc647859':
ok_genes.append(gene_row)
ok_gnames.append(genes[idx])
print 'Selected genes: ' + str(len(ok_gnames))
for idx, gene in enumerate(ok_gnames):
if len(gdict[gene])==1:
chr = '0'+ gdict[gene]
else:
chr = gdict[gene]
ok_gnames[idx]= chr + ' ' + gene
ok_genes=array(ok_genes)
r.library('gplots')
colors = r.redgreen(len(unique(ok_genes)))
colors = colors[::-1]
#colors[2]='#880000'
colors[3]='#880000'
#return ok_genes, samples, ok_gnames, colors
cfnames.append('clustergram_' + str(t).replace('.','') + '_' + str(len(ok_gnames)) + '.png')
hfnames.append('heatmap_' + str(t).replace('.','') + '_' + str(len(ok_gnames)) + '.png')
callRforClustergram(cfnames[-1], ok_genes, samples, ok_gnames, colors)
makeHeatmap(hfnames[-1], ok_genes, samples, ok_gnames, colors)
return hstack([cfnames, hfnames])
PennCNV
Runs PennCNV to detect CNV regions
Output files: pglfeb.csv_11s.tab.out , pglfeb.csv_11t.tab.out , pglfeb.csv_12s.tab.out , pglfeb.csv_12t.tab.out , pglfeb.csv_13s.tab.out , pglfeb.csv_13t.tab.out , pglfeb.csv_14Bt.tab.out , pglfeb.csv_14s.tab.out , pglfeb.csv_19s.tab.out , pglfeb.csv_19t.tab.out , pglfeb.csv_1s.tab.out , pglfeb.csv_1t.tab.out , pglfeb.csv_20s.tab.out , pglfeb.csv_20t.tab.out , pglfeb.csv_21s.tab.out , pglfeb.csv_21t.tab.out , pglfeb.csv_2s.tab.out , pglfeb.csv_2t.tab.out , pglfeb.csv_32s.tab.out , pglfeb.csv_32t.tab.out , pglfeb.csv_33s.tab.out , pglfeb.csv_33t1.tab.out , pglfeb.csv_34s.tab.out , pglfeb.csv_34t.tab.out , pglfeb.csv_36s.tab.out , pglfeb.csv_36t.tab.out , pglfeb.csv_37s.tab.out , pglfeb.csv_37t.tab.out , pglfeb.csv_3s.tab.out , pglfeb.csv_3t.tab.out , pglfeb.csv_43s.tab.out , pglfeb.csv_43t.tab.out , pglfeb.csv_44s.tab.out , pglfeb.csv_44t.tab.out , pglfeb.csv_45s.tab.out , pglfeb.csv_45t.tab.out , pglfeb.csv_4s.tab.out , pglfeb.csv_4t.tab.out , pglfeb.csv_5Bt.tab.out , pglfeb.csv_5s.tab.out , pglfeb.csv_5t.tab.out , pglfeb.csv_6s.tab.out , pglfeb.csv_6t.tab.out , pglfeb.csv_7s.tab.out , pglfeb.csv_7t.tab.out , pglfeb.csv_8s.tab.out , pglfeb.csv_8t.tab.out .
Last build was: Sun Jan 8 19:10:54 2012.
Required CPU time: 00:52:42.29.
def PennCNV(inputfiles):
"""Runs PennCNV to detect CNV regions"""
ofiles = list()
for ifile in inputfiles:
cmdstr = 'perl ' + penncnv_path + \
'/detect_cnv.pl -test -hmm ' + \
penncnv_path + '/lib/hhall.hmm -pfb ' + \
penncnv_path + '/lib/ho1v1.hg18.pfb -gcmodelfile ' + \
penncnv_path + '/lib/ho1v1.hg18.gcmodel ' + \
ifile + \
' -log "' + ifile + '.log" ' + \
'-out "' + fname + '_' + ifile + '.out"'
print 'Executing: ' + cmdstr
#print 'WARNING: skipping PennCNV call! Uncomment to really call.'
os.system(cmdstr)
#ofiles.append(lp.newFile(fname + '_' + ifile + '.out'))
ofiles.append(fname + '_' + ifile + '.out')
return ofiles
getSampleNames
Extracts sample names
Last build was: Sun Jan 8 03:47:10 2012.
Required CPU time: 00:00:0.03.
def getSampleNames(fname):
"""Extracts sample names"""
b=open(fname).readlines()
t=list()
p = getPairedSampleIDs()
for l in b[1:]:
s = l.split()[0]
#if s in p:
t.append(s)
return t
CompareRegionAndGenes
Computes intersections between regions and genes
Last build was: Sun Jan 8 03:47:09 2012.
Required CPU time: 00:00:13.22.
def CompareRegionAndGenes(data):
"""Computes intersections between regions and genes"""
ref = open(penncnv_path+'/ucsc_hg18/refGene.txt').readlines()
for idx, line in enumerate(ref):
ref[idx] = line.split()
refgenes = [ref[i][12] for i in range(0, len(ref))]
res = list()
for idx, line in enumerate(data[1:]):
if mod(idx, 100)==0:
print 'Intersecting: '+str(idx)+' of '+str(len(data)-1)
line = line.split('\t')
gene = line[10]
try:
where = refgenes.index(gene)
int1 = regIntersect([int(line[1]), int(line[2])], [int(ref[where][4]), int(ref[where][5])])
int2 = regIntersect([int(line[1]), int(line[2])], [int(ref[where][6]), int(ref[where][7])])
#res.append(['***********************************************'])
res.append([
gene,
line[6], #sample
line[7], #s/t
int(line[1]), #reg start
int(line[2]), #reg end
int(line[2])-int(line[1]), # reg size
int(ref[where][4]), #trans start
int(ref[where][5]), #trans end
int(ref[where][5])-int(ref[where][4]),
int(ref[where][6]), #code start
int(ref[where][7]), #code end
int(ref[where][7])-int(ref[where][6]),
int1, #int trans
int2, #int code
])
except:
res.append([
gene,
line[6],
line[7],
int(line[1]),
int(line[2]),
'?', '?', '?', '?', '?', '?','?','?'
])
return res
exportFigExtRegs
Creates a figure for the extended regions using R
Output file: extRegs.pdf
Last build was: Sun Jan 8 19:45:02 2012.
Required CPU time: 00:00:3.74.
def exportFigExtRegs(frags):
"""Creates a figure for the extended regions using R"""
r.library('quantsmooth')
chrwidth = 10
chrspacing = 30
r.pdf(file='extRegs.pdf', height=10, width=12)
#maxarmlen = max(
maxlen = max([a[0][1] for a in frags['1']])
r.plot(1,type="n",xlim=[0,(chrspacing+chrwidth)*(len(frags)-1)],ylim=[0, maxlen],xaxt="n",yaxt="n", ann=False, frame_plot=False, tick_number=0, xlab="Autosomes")
keys = [int(k) for k in frags]
skeys = sort(keys)
tickat = list()
for cidx,chrom in enumerate([str(k) for k in skeys]):
offs = cidx*(chrspacing+chrwidth)
#maxy = max([a[0][1] for a in frags[chrom]])
tothits = sum([a[0][1]-a[0][0] for a in frags[chrom][0::2]])
maxlen = max([a[0][1] for a in frags[chrom]])
maxy = r.lengthChromosome(chrom, units = 'bases')[chrom]
r.lines([offs, offs], [0,maxy])
for fidx, frag in enumerate(frags[chrom]):
if frag[1]=='' and frag[2]=='':
continue
y = (frag[0][0]+float(frag[0][1]))/2
y = (y * maxy) / maxlen
l = frag [0][1]-frag[0][0]+1
lB = int(frag[1])
lT = int(frag[2])
Ts = sum([a=='T' for a in frag[3]])
Bs = sum([a=='B' for a in frag[3]])
#Ts_p = sum([a=='T' for idx,a in enumerate(frag[3]) if int(frag[4][idx])>2])
#Ts_n = sum([a=='T' for idx,a in enumerate(frag[3]) if int(frag[4][idx])<2])
#Bs_p = sum([a=='B' for idx,a in enumerate(frag[3]) if int(frag[4][idx])>2])
#Bs_n = sum([a=='B' for idx,a in enumerate(frag[3]) if int(frag[4][idx])<2])
Ts_p = sum([a=='T' for idx,a in enumerate(frag[3]) if int(frag[4][idx])>2])
Ts_n = sum([a=='T' for idx,a in enumerate(frag[3]) if int(frag[4][idx])<2])
Bs_p = sum([a=='B' for idx,a in enumerate(frag[3]) if int(frag[4][idx])>2])
Bs_n = sum([a=='B' for idx,a in enumerate(frag[3]) if int(frag[4][idx])<2])
Tgain = Ts_p - Ts_n
Bgain = Bs_p - Bs_n
#Tcol = 'red' if Tgain < 0 else 'green'
#Bcol = 'red' if Bgain < 0 else 'green'
Dcol = 'darkred' if Tgain - Bgain > 0 else 'darkgreen'
armlen = int(abs(lT - lB))*1.5
if max(lB, lT)>0:
if Dcol == 'darkred':
r.lines([offs +chrwidth-2 , armlen + offs +chrwidth-2], [maxy-y,maxy-y], col=Dcol)
else:
r.lines([offs -armlen -chrwidth+2, offs -chrwidth+2], [maxy-y,maxy-y], col=Dcol)
tickat.append(offs)
print chrom, tothits/float(maxy)
bleachv = 1-min((tothits/float(maxy))/0.08,1)
r.paintCytobands(chrom, pos=[offs+chrwidth/2, maxy], width=chrwidth, units="bases", orientation='v', legend=False, bleach=bleachv)
# r.axis(at=tickat, labels=range(1,23), cex_axis=1, side=range(1,23), mgp=[3,.5,0], xlab='Autosomes', main='Genomic regions affected by CNV')
r.axis(at=tickat, lty=0, labels=range(1,23), cex_axis=1, side=range(1,23), xlab='Autosomes', main='Genomic regions affected by CNV', mgp=[3,-0.8,0])
# if Bgain:
# #r.lines([-log10(l) + offs, 0 + offs], [y,y], col=Bcol)
# r.lines([offs - lB -1, 0 + offs -1], [y,y], col=Bcol)
# r.lines([offs - lB -1, 0 + offs -1], [y,y], col=Bcol)
# if Tgain:
# #r.lines([0 + offs, log10(l) + offs], [y,y], col=Tcol)
# r.lines([0 + offs +1 , lT + offs +1], [y,y], col=Tcol)
# r.lines([0 + offs +1 , lT + offs +1], [y,y], col=Tcol)
r.dev_off()
return('extRegs.pdf')
manualClean
Removes a list of CNVi-related genes produced elsewhere
Last build was: Sun Jan 8 03:46:55 2012.
Required CPU time: 00:00:3.48.
def manualClean(x):
"""Removes a list of CNVi-related genes produced elsewhere"""
no=[int(n) for n in open('manuallyRemovedCNVs.txt').read().split()]
cleandata = list()
f = open('cleanedCNVs.txt', 'w')
for idx, line in enumerate(x):
if not (idx+1 in no):
cleandata.append(line)
f.write(line)
return cleandata
samplesDistMats
Computes Tanimoto distance between samples
Last build was: Sun Jan 8 03:47:21 2012.
Required CPU time: 00:00:0.24.
def samplesDistMats(dataset):
"""Computes Tanimoto distance between samples"""
inters_mat = squareform(pdist(dataset, lambda x,y: sum(logical_and(x,y))))
for idx in range(0,dataset.shape[0]):
inters_mat[idx,idx]=sum(dataset,axis=1)[idx]
notIntersMat = squareform(pdist(dataset, lambda x,y: sum(~logical_and(x,y))))
tanimotoMat = squareform(pdist(dataset, lambda x,y: 1 - sum(logical_and(x,y))/float(sum(logical_or(x,y))) ))
return [inters_mat, notIntersMat, tanimotoMat]
exportPerRegion_clean
Dummy node to skip manual filtering phase
Output file: perRegion_clean.csv
Last build was: Sun Jan 8 19:45:02 2012.
Required CPU time: 00:00:0.14.
def exportPerRegion_clean(data):
"""Dummy node to skip manual filtering phase"""
f = open('perRegion_clean.csv','w')
sdict = idToNameDict()
f.write(data[0].replace('\t',',')+'\n')
for line in data[1:]:
l = line.replace(',','.').split()
l[6] = sdict[l[6]]
l = str(l).replace('\'', '').strip('[]')
f.write(l+'\n')
return 'perRegion_clean.csv'
exportCNVDiffMat
Exports to tsv file
Output file: CNVgenes.csv
Last build was: Sun Jan 8 19:45:15 2012.
Required CPU time: 00:00:12.21.
def exportCNVDiffMat(cnvdiff, cnvdata, fisher, genes, samples):
"""Exports to tsv file"""
p = getPairings()
samples = list()
for a in p:
samples.extend([a[0], a[1]])
samples = samplesToNames(samples)
data = list()
for line in cnvdata:
data.append(line.split('\t'))
data = array(data)
vects, vs, vt = cnvdiff[0], cnvdiff[1], cnvdiff[2]
gdict = geneChrDict()
for i in range(len(samples),0,-1):
if mod(i,2)==0:
samples.insert(i, samples[i-1]+'-'+samples[i-2])
header =['gene', 'Chr',
'B gain', 'B loss', 'B CNV',
'T gain', 'T loss', 'T CNV',
'B CNV + T CNV',
'TvsB loss','TvsB gain', 'TvsBgain+TvsBloss', 'TvsB loss/gain p-value',
'max CNV', 'Reg. Max length',
'CNV samples (repeated if hit in multiple regions)']
header = hstack([header, samples])
fisher_abs = fisher[0]
fisher_diff = fisher[1]
f = open('CNVgenes.csv', 'w')
f.write(str(header).strip('[]').replace('\n','').replace('\' \'',',').strip('\'') + '\n')
for idx, vect in enumerate(vects):
#gene name
f.write(genes[idx])
#if genes[idx]=='ACOT11':
# import pdb; pdb.set_trace()
#chromosome
f.write(',' + str(gdict[genes[idx]]))
#cnv stats
cnvBgain = sum(vs[idx]>2)
cnvBloss = sum(vs[idx]<2)
cnvB = cnvBgain + cnvBloss
cnvTgain = sum(vt[idx]>2)
cnvTloss = sum(vt[idx]<2)
cnvT = cnvTgain + cnvTloss
f.write(', ' + str(cnvBgain) + ',' + str(cnvBloss) + ',' + str(cnvB))
f.write(', ' + str(cnvTgain) + ',' + str(cnvTloss) + ',' + str(cnvT))
f.write(', ' + str(cnvB+cnvT))
# f.write(', ' + str(cnvB) + ',' + str(cnvT) + ',' + str(cnvB+cnvT))
#fisher on B vs T cnvs
#f.write(','+str(fisher_abs[idx]))
#differential cnvs stats
f.write(str(',' +
str(sum(vect<0))+','+
str(sum(vect>0))+','+
str(sum(vect<0)+
sum(vect>0))+',')
.replace('\n',''))
#fisher on s vs t differential cnvs
f.write(',' + str(fisher_diff[idx]) + ',')
# max CNV
cns = hstack([vs[idx],vt[idx]])
maxcnv = cns[argmax(abs(2-cns))]
f.write(str(maxcnv) + ',')
# per gene additional info
info = extractPerGeneProperties(data, genes[idx])[1:]
for idx2, item in enumerate(info):
if not item=='nan':
if idx2 in [3,4,5]:
f.write(item.replace(',','').replace('\n','').replace('\'', '').replace('[','').replace(']','')+',')
#cnv per sample
f.write(str(zip(vs[idx],vt[idx],vect)).strip('[]').replace('(','').replace(')','').replace('\n','').replace('.0',''))
f.write('\n')
return 'CNVgenes.csv'
exportMergedFragments
Exports extended regions to tsv file
Output file: extRegions.tsv
Last build was: Sun Jan 8 19:45:15 2012.
Required CPU time: 00:00:0.09.
def exportMergedFragments(mf):
'''Exports extended regions to tsv file'''
f = open('extRegions.tsv', 'w')
f.write('Chr.\t'+
'ExtReg. Start\t'+
'ExtReg. End\t'+
'ExtReg. Length\t'+
'Max hits B\t'+
'Max hits T\t'+
'B/T\t'+
'CN\t'+
'"Genes"\n')
for chrom in mf:
for extreg in mf[chrom]:
f.write(
#chromosome
chrom+'\t'+
#reg start
str(extreg[0][0])+'\t'+
#reg end
str(extreg[0][1])+'\t'+
#reg length
str(extreg[0][1]-extreg[0][0]+1)+'\t'+
# max hits B
str(extreg[1])+'\t'+
# max hits T
str(extreg[2])+'\t'+
#BT
str(extreg[3]).strip('[]').replace("'",'').replace(',','')+'\t'+
#CN
str(extreg[4]).strip('[]').replace("'",'').replace(',','')+'\t'+
#genes
str(extreg[5]).replace('set','').replace('(','').replace(')','')
.replace('[','').replace(']','').replace("'",'')+'\n'
)
return 'extRegions.tsv'
exportMergedRegsTB
Exports to tsv file
Output file: mergedRegsTB.tsv
Last build was: Sun Jan 8 19:45:16 2012.
Required CPU time: 00:00:0.66.
def exportMergedRegsTB(mergedRegs):
"""Exports to tsv file"""
f = open('mergedRegs.tsv', 'w')
f.write('Chr.\tReg. Start\tReg. End\tReg. Lenght\tNum Samples\tCN-B\tCN-T\tnB\tnT\t"Genes"\n')
for key in sorted(mergedRegs.keys()):
for i in range(0, len(mergedRegs[key][0])):
# try:
try:
cnb = [cn for idx, cn in enumerate(mergedRegs[key][3][i])
if mergedRegs[key][4][i][idx]=='B']
cnt = [cn for idx, cn in enumerate(mergedRegs[key][3][i])
if mergedRegs[key][4][i][idx]=='T']
except:
import pdb; pdb.set_trace()
f.write(
# Chr
key+'\t'+
#Reg. Start e Reg. End
str(mergedRegs[key][0][i]).strip('[]').replace(', ','\t')+'\t'+
#Reg. Length
str(mergedRegs[key][0][i][1]-mergedRegs[key][0][i][0]+1)+'\t'+
#Num Samples
str(mergedRegs[key][1][i])+'\t'+
#CN-B
str(cnb).
replace('[','').
replace(']','').
replace("'", '')+'\t'+
#CN-T
str(cnt).
replace('[','').
replace(']','').
replace("'", '')+'\t'+
#nB
str(sum([st=='B' for st in mergedRegs[key][4][i]]))+'\t'+
#nT
str(sum([st=='T' for st in mergedRegs[key][4][i]]))+'\t'+
#Genes
str(mergedRegs[key][2][i]).replace('set','').
replace(')','').
replace('(','').
replace('[','').
replace(']','').
replace("'", '')+'\n'
)
# except: import pdb; pdb.set_trace()
return 'mergedRegsTB.tsv'
mergeFragments
Merges all contiguous non-void region fragments
Last build was: Sun Jan 8 19:44:58 2012.
Required CPU time: 00:00:0.37.
def mergeFragments(intRegs):
"""Merges all contiguous non-void region fragments"""
extreg=dict()
for chrom in intRegs:
extreg[chrom] = list()
currExtRegBT = list()
currExtRegCN = list()
currExtRegS = [Inf,0]
currExtRegG = set()
currExtRegMaxhitsB = 0
currExtRegMaxhitsT = 0
for idx, frag in enumerate(intRegs[chrom][4]):
a = intRegs[chrom][0][idx][0]
b = intRegs[chrom][0][idx][1]
#import pdb; pdb.set_trace()
if frag != []:
currExtRegMaxhitsT = max(currExtRegMaxhitsT, sum([a1=='T' for a1 in frag]))
currExtRegMaxhitsB = max(currExtRegMaxhitsB, sum([a1=='B' for a1 in frag]))
currExtRegBT.extend(frag)
currExtRegCN.extend(intRegs[chrom][3][idx])
currExtRegS = [min(currExtRegS[0], a), max(currExtRegS[1],b)]
currExtRegG = currExtRegG.union(intRegs[chrom][2][idx])
else:
extreg[chrom].append([currExtRegS, currExtRegMaxhitsB,
currExtRegMaxhitsT, currExtRegBT, currExtRegCN, currExtRegG])
extreg[chrom].append([[a,b], '', '', '', '', 'unknown'])
currExtRegCN=list()
currExtRegBT=list()
currExtRegG = set()
currExtRegS = [Inf,0]
currExtRegMaxhitsT = 0
currExtRegMaxhitsB = 0
# handle last fragment (will get no '[]' as stop signal)
if frag != []:
extreg[chrom].append([currExtRegS, currExtRegMaxhitsB,
currExtRegMaxhitsT, currExtRegBT, currExtRegCN, currExtRegG])
return extreg
computeFisher
Tests the hyphotesis that a gene is hit by CNV in M samples out of N by chance
Last build was: Sun Jan 8 19:43:52 2012.
Required CPU time: 00:00:8.96.
def computeFisher(cnvect):
"""Tests the hyphotesis that a gene is hit by CNV in M samples out of N by chance"""
svec = cnvect[1]
tvec = cnvect[2]
diff = cnvect[0]
numsamp = len(svec[0])
fishval_diff = zeros(svec.shape[0])
fishval_abs = zeros(svec.shape[0])
for idx, row in enumerate(diff):
a11 = sum(row < 0)
a12 = numsamp-a11
a21 = sum(row > 0)
a22 = numsamp-a21
#fishval[idx]=r.fisher_test(array([[a11,a12],[a21,a22]]), alternative='less')['p.value']
fishval_diff[idx]=r.fisher_test(array([[a11,a12],[a21,a22]]))['p.value']
for idx, row in enumerate(svec):
a11 = sum(row != 2)
a12 = numsamp-a11
a21 = sum(tvec[idx] != 2)
a22 = numsamp-a21
fishval_abs[idx]=r.fisher_test(array([[a11,a12],[a21,a22]]))['p.value']
return fishval_abs, fishval_diff
prepareInput
Converts data to a format suitable for PennCNV
Output files: 11s.tab , 11t.tab , 12s.tab , 12t.tab , 13s.tab , 13t.tab , 14Bt.tab , 14s.tab , 19s.tab , 19t.tab , 1s.tab , 1t.tab , 20s.tab , 20t.tab , 21s.tab , 21t.tab , 2s.tab , 2t.tab , 32s.tab , 32t.tab , 33s.tab , 33t1.tab , 34s.tab , 34t.tab , 36s.tab , 36t.tab , 37s.tab , 37t.tab , 3s.tab , 3t.tab , 43s.tab , 43t.tab , 44s.tab , 44t.tab , 45s.tab , 45t.tab , 4s.tab , 4t.tab , 5Bt.tab , 5s.tab , 5t.tab , 6s.tab , 6t.tab , 7s.tab , 7t.tab , 8s.tab , 8t.tab .
Last build was: Sun Jan 8 03:42:33 2012.
Required CPU time: 00:04:57.23.
def prepareInput(fname, ssheet):
"""Converts data to a format suitable for PennCNV"""
ss = [l.split('\t') for l in open(ssheet).readlines()][1:]
scolumns = range(4,200,3)
#a=open(fname).readline()
#samples = [line.replace('.GType','') for line in a.split(',')[3::3]]
samples = [s[0] for s in ss]
fnames = list()
for k, sample in enumerate(samples):
print 'Preparazione campione ' + sample + '...',
#print 'SKIPPING!'
os.system('cut -f 1-3,' + str(scolumns[k]) + '-' + str(scolumns[k]+2) + ' --delimiter , "' + fname +'" > ' + sample + '.csv')
os.system('cat ' + sample + ".csv | tr ',' '\t' > " + sample + '.tab')
os.system('rm ' + sample + '.csv')
fnames.append(sample + '.tab')
return fnames
exportFigCNVlenghts
Creates a figure of the samples based % of genome affected by CNV
Output files: cnvlenghts.pdf , cnvlenghts_log.pdf .
Last build was: Sun Jan 8 19:45:16 2012.
Required CPU time: 00:00:0.04.
def exportFigCNVlenghts(countCNVlenghts_o):
"""Creates a figure of the samples based % of genome affected by CNV"""
r.pdf('cnvlenghts_log.pdf')
y = [int(x[1])/1000000 for x in countCNVlenghts_o if not 'B' in x[0]]
l = [x[0] for x in countCNVlenghts_o if not 'B' in x[0]]
yidx = sorted(range(len(y)), key = y.__getitem__)
ys = sorted(y)
ls = [l[x] for x in yidx]
r.barplot(ys, horiz=True, names_arg=ls, log='x', cex_names=0.5, las=1)
r.dev_off()
r.pdf('cnvlenghts.pdf')
y = [int(x[1]) for x in countCNVlenghts_o if not 'B' in x[0]]
l = [x[0] for x in countCNVlenghts_o if not 'B' in x[0]]
yidx = sorted(range(len(y)), key = y.__getitem__)
ys = sorted(y)
ls = [l[x] for x in yidx]
r.barplot(ys, horiz=True, names_arg=ls, cex_names=0.5, las=1)
#r.lines(range(0,len(ys)), r.cumsum(ys))
#r.lines(r.cumsum(y), range(0,len(y)))
r.dev_off()
return ('cnvlenghts.pdf', 'cnvlenghts_log.pdf')
makeBed
Create Bed file for use with Genome Browser
Output file: pglfeb.csv.bed
Last build was: Sun Jan 8 19:45:16 2012.
Required CPU time: 00:00:0.42.
def makeBed(CNVinput):
"""Create Bed file for use with Genome Browser"""
os.system('perl ' + penncnv_path + '/visualize_cnv.pl -format bed -output "' + fname + '.bed" "' + CNVinput + '"')
return fname + '.bed'
CNVDiffMat
Makes a matrix of B/T difference in CN between same genes of different samples
Last build was: Sun Jan 8 19:43:41 2012.
Required CPU time: 00:00:2.43.
def CNVDiffMat(fname, genes, allsamples):
"""Makes a matrix of B/T difference in CN between same genes of different samples"""
data=list()
f=fname
for line in f:
line=line[0:-1]
data.append(line.split('\t'))
data=array(data)
hashd = dict( zip( genes, range(0,len(genes)) ) )
p = getPairings()
sp = [x[0] for x in p]
tp = [x[1] for x in p]
s = list()
for sample in sp:
for line in data:
if sample == line[6]:
s.append(line)
t = list()
for sample in tp:
for line in data:
if sample == line[6]:
t.append(line)
samples = sp
s_vectors = np.zeros((len(samples),len(genes)))*nan
s_vectors_min =np.zeros((len(samples),len(genes)))*nan
s_vectors_num =np.zeros((len(samples),len(genes)))*nan
s_vectors_cn = np.zeros((len(samples),len(genes)))*nan
for s_idx, sample in enumerate(samples):
print s_idx, 'of', len(samples),',',
x = nonzero([l[6]==sample for l in s])[0]
sub_s = [s[pickme] for pickme in x]
sub_s = array(sub_s)
sample_genes = sub_s[:,10]
for gene in unique(sample_genes):
if isnan(s_vectors[s_idx][hashd[gene]]):
temp = array(sub_s[sub_s[:,10]==gene][:,5], int)
maxidx = argmax(temp)
minidx = argmin(temp)
absmax = temp[maxidx]
absmin = temp[minidx]
s_vectors[s_idx][hashd[gene]] = absmax
s_vectors_min[s_idx][hashd[gene]] = absmin
s_vectors_cn[s_idx][hashd[gene]] = assignGeneCN(temp)
s_vectors_num[s_idx][hashd[gene]] = len(temp)
print
s_vectors[isnan(s_vectors)] = 2
s_vectors_min[isnan(s_vectors_min)] = 2
s_vectors_cn[isnan(s_vectors_cn)] = 2
s_vectors_num[isnan(s_vectors_num)] = 0
samples = tp
t_vectors = np.zeros((len(samples),len(genes)))*nan
t_vectors_min = np.zeros((len(samples),len(genes)))*nan
t_vectors_num =np.zeros((len(samples),len(genes)))*nan
t_vectors_cn = np.zeros((len(samples),len(genes)))*nan
for t_idx, sample in enumerate(samples):
print t_idx, 'of', len(samples),',',
x = nonzero([l[6]==sample for l in t])[0]
sub_t = [t[pickme] for pickme in x]
sub_t = array(sub_t)
try:
sample_genes = sub_t[:,10]
except IndexError:
import pdb; pdb.set_trace()
for gene in unique(sample_genes):
#if gene == 'PC':
# import pdb; pdb.set_trace()
if isnan(t_vectors[t_idx][hashd[gene]]):
temp = array(sub_t[sub_t[:,10]==gene][:,5], int)
maxidx = argmax(temp)
absmax = temp[maxidx]
minidx = argmin(temp)
absmin = temp[minidx]
t_vectors[t_idx][hashd[gene]] = absmax
t_vectors_min[t_idx][hashd[gene]] = absmin
t_vectors_num[t_idx][hashd[gene]] = len(temp)
t_vectors_cn[t_idx][hashd[gene]] = assignGeneCN(temp)
print
t_vectors[isnan(t_vectors)]=2
t_vectors_min[isnan(t_vectors_min)] = 2
t_vectors_cn[isnan(t_vectors_cn)] = 2
t_vectors_num[isnan(t_vectors_num)] = 0
return [t_vectors_cn.T-s_vectors_cn.T, #0
s_vectors_cn.T, t_vectors_cn.T, #1,2
s_vectors_min.T, t_vectors_min.T, #3,4
s_vectors_num.T, t_vectors_num.T, #5,6
s_vectors.T, t_vectors.T] #7,8
exportComRegGen
Exports CompareRegionAndGenes output to tsv formatOutput file: CompareRegGenes.txt
Last build was: Sun Jan 8 03:47:10 2012.
Required CPU time: 00:00:0.08.
def exportComRegGen(data):
"""Exports CompareRegionAndGenes output to tsv format"""
f = open('CompareRegGenes.txt','w')
f.write('Gene\tSample\ts/t\tReg. start\tReg. end\tReg. size\tTanscript. start\t Transcript. end\tTranscript. size\tCode start\t Code end\tCode size\tInters. to Transcr. %\t Inters. to Code %')
for line in data:
if line[0]!='NOT_FOUND':
for item in line:
if type(item)==float:
f.write(str(item)+'\t')
f.write('\n')
return 'CompareRegGenes.txt'
exportMergedRegs
Exports to tsv fileOutput file: mergedRegsTB.tsv
Last build was: Sun Jan 8 03:47:16 2012.
Required CPU time: 00:00:0.13.
def exportMergedRegs(mergedRegs):
"""Exports to tsv file"""
f = open('mergedRegsTB.tsv', 'w')
f.write('B/T\tChr\tReg. Start\tReg. End\tNum Samples\tCN\t"Genes"\n')
for key in sorted(mergedRegs.keys()):
for i in range(0, len(mergedRegs[key][0])):
try:
f.write(('B' if key[0] == 's' else 'T') +'\t'+
key[1]+'\t'+
str(mergedRegs[key][0][i]).strip('[]').replace(', ','\t')+'\t'+
str(mergedRegs[key][1][i])+'\t'+
str(mergedRegs[key][3][i]).
replace('[','').
replace(']','').
replace("'", '')+'\t'+
str(mergedRegs[key][2][i]).replace('set','').
replace(')','').
replace('(','').
replace('[','').
replace(']','').
replace("'", '')+'\n'
)
except: import pdb; pdb.set_trace()
return 'mergedRegsTB.tsv'
exportIntersMat_genes
Exports the output of intersMat_genes to csv formatOutput file: verbose_inters_mat.txt
Last build was: Sun Jan 8 03:47:20 2012.
Required CPU time: 00:00:0.08.
def exportIntersMat_genes(genes, samples, inters_names):
"""Exports the output of intersMat_genes to csv format"""
f = open('verbose_inters_mat.txt', 'w')
f.write(' ,'+','.join(map(lambda x:x[0]+x[1], samples)) + '\n')
for idx1 in range(0,len(inters_names)):
f.write(samples[idx1][0]+samples[idx1][1]+',')
for idx2 in range(0,len(inters_names)):
f.write(str(list(inters_names[idx1][idx2])).strip('[]').replace('\'','').replace(',','') + ',')
f.write('\n')
return 'verbose_inters_mat.txt'
joinPennCNVout
Puts together the different ouptuts (1 per sample) of PennCNVOutput file: pglfeb.csv_allNO14.out
Last build was: Sun Jan 8 03:45:59 2012.
Required CPU time: 00:00:0.11.
def joinPennCNVout(inputfiles):
"""Puts together the different ouptuts (1 per sample) of PennCNV"""
#log_ofile = open(fname + '_all.log', 'a')
if os.path.exists(fname + '_allNO14.out'):
os.remove(fname+'_allNO14.out')
out_ofile = open(fname + '_allNO14.out', 'a')
for ifile in inputfiles:
#log_ifname = ifile + '.log'
#log_ofile.write(open(log_ifname, 'r').read())
#if not '14B' in ifile and not '5B' in ifile and not '34' in ifile and not '33' in ifile:
#if not '14' in ifile and not '21' in ifile and not '14B' in ifile and not '5B' in ifile:
out_ofile.write(open(ifile, 'r').read())
out_ofile.close()
return fname + '_allNO14.out'
exportPerRegion
Exports to tsv fileOutput file: perRegion.csv
Last build was: Sun Jan 8 03:51:58 2012.
Required CPU time: 00:00:0.26.
def exportPerRegion(data):
"""Exports to tsv file"""
f = open('perRegion.csv','w')
sdict = idToNameDict()
f.write(data[0].replace('\t',',')+'\n')
for line in data[1:]:
l = line.replace(',','.').split()
l[6] = sdict[l[6]]
l = str(l).replace('\'', '').strip('[]')
f.write(l+'\n')
return 'perRegion.csv'
addGeneInfo
Annotate regions with intersecting genesOutput file: pglfeb.csv_allNO14.out.genes
Last build was: Sun Jan 8 03:46:51 2012.
Required CPU time: 00:00:51.95.
def addGeneInfo(fname):
"""Annotate regions with intersecting genes"""
#print 'SKIPPING'
#return fname + '.genes'
os.system('perl ' + penncnv_path + '/scan_region.pl "' + fname + '" -refgene ' + annot_path + '/refGene.txt -reflink ' + annot_path + '/refLink.txt > "' + fname + '.genes1"')
os.system('perl ' + penncnv_path + '/scan_region.pl "' + fname + '.genes1" -refgene ' + annot_path + '/refGene.txt -reflink ' + annot_path + '/refLink.txt -expandleft 5m > "' + fname + '.genes2"')
os.system('perl ' + penncnv_path + '/scan_region.pl "' + fname + '.genes2" -refgene ' + annot_path + '/refGene.txt -reflink ' + annot_path + '/refLink.txt -expandright 5m > "' + fname + '.genes"')
os.system('rm "' + fname + '".genes1')
os.system('rm "' + fname + '".genes2')
#os.system('perl ' + penncnv_path + '/detect_cnv.pl -cctest -pfb ' + penncnv_path + '/lib/ho1v1.hg18.pfb -phenofile phenotype -cnv "' + fname + '" > "' + fname + '".CC')
return fname + '.genes'
sampleSheet
The name of the filename containing sample IDs and namesOutput file: sample_sheet.tsv
Last build was: Sun Jan 8 03:37:36 2012.
Required CPU time: 00:00:0.03.
def sampleSheet():
"""The name of the filename containing sample IDs and names"""
return('sample_sheet.tsv')
readFile
From now on the input does not come from filesLast build was: Sun Jan 8 03:46:51 2012.
Required CPU time: 00:00:0.06.
def readFile(fname):
"""From now on the input does not come from files"""
return open(fname).readlines()
distMatGfx
Produces an MDS visualization of the output of samplesDistMatsOutput files: t_tani_distrib.pdf , t_tani_mds.pdf .
Last build was: Sun Jan 8 03:51:56 2012.
Required CPU time: 00:00:0.25.
def distMatGfx(samples, mats):
"""Produces an MDS visualization of the output of samplesDistMats"""
tani = mats[2]
test_couples=[['13t','14t'], ['33t1','33t2'], ['34t','34tL'], ['5t', '5Bt']]
#s = getSampleNames()
s = samples
tani_t = list()
tani_t_mat = list()
tids = list()
print 'Adding background distances :'
for idx1, s1 in enumerate(s):
matrow = list()
if 't' in s1:
tids.append(s1)
for idx2, s2 in enumerate(s):
if 't' in s2:
matrow.append(tani[idx1,idx2])
if s1 != s2:
tani_t.append(tani[idx1,idx2])
#print s[idx1] + '-' + s[idx2]+',',
tani_t_mat.append(matrow)
#hist(tani_t,30)
test_dists=list()
sdict = idToNameDict()
for idx, item in enumerate(test_couples):
if item[0] not in s or item[1] not in s:
continue
s1 = s.index(item[0])
s2 = s.index(item[1])
d = tani[s1,s2]
test_dists.append(d)
#annotate('('+sdict[item[0]]+','+sdict[item[1]]+')', [d,10], [d,10*idx+20])
print d
mds=r.cmdscale(array(tani_t_mat))
#savefig('t_tani_distrib.pdf')
#figure()
#plot(mds[:,0], mds[:,1], '.')
for idx, item in enumerate(tids):
pass
#annotate(sdict[item], [mds[idx,0], mds[idx,1]])
#savefig('t_tani_mds.pdf')
return ['t_tani_distrib.pdf', 't_tani_mds.pdf']
intersectTBRegs
Intersects all CNV T regions against all CNV B regionsLast build was: Sun Jan 8 19:44:57 2012.
Required CPU time: 00:00:7.36.
def intersectTBRegs(getSampleNames, manualClean):
"""Intersects all CNV T regions against all CNV B regions"""
f,h = PennCNVParser()
manualClean = [f.split(l) for l in manualClean]
num = dict()
num['s'] = sum(['s' in s for s in getSampleNames])
num['t'] = sum(['t' in s for s in getSampleNames])
regsets = dict()
chrs = set([l[1] for l in manualClean])
for samp in getSampleNames:
for chrom in chrs:
sub = [l for l in manualClean if l[7]==samp and l[1] == chrom]
st = 'B' if 's' in samp else 'T'
regset = [[int(l[2]),int(l[3]), l[10], l[6], st] for l in sub]
if not chrom in regsets.keys():
regsets[chrom] = list()
if not sub == []:
regsets[chrom].append(regset)
a = dict()
for chrom in chrs:
# if chrom == '13':
# import pdb; pdb.set_trace()
a[chrom]=intersRegs([sorted(s) for s in regsets[chrom]])
return a
exportToCircos
Exports to a file suitable for use with CircosLast build was: Sun Jan 8 03:51:59 2012.
Required CPU time: 00:00:0.02.
def exportToCircos(samples, x):
"""Exports to a file suitable for use with Circos"""
return "tobefixed!"
x=[l.split('\t') for l in x][1:]
x=[[l[0],l[1],l[2],l[5],l[6]] for l in x]
fnames = list()
for sample in samples:
fname = 'circosInput_' + sample + '.txt'
fnames.append(fname)
f = open(fname, 'w')
subx = [l[0:4] for l in x if l[4]==sample]
f.writelines("hs%s\n" % str(item).strip('[]').replace('\'','').replace(',','') for item in subx)
return(fnames)
intersectRegs
Intersects all CNV regions of T and B samples separatelyLast build was: Sun Jan 8 03:47:15 2012.
Required CPU time: 00:00:5.19.
def intersectRegs(getSampleNames, manualClean):
"""Intersects all CNV regions of T and B samples separately"""
f,h = PennCNVParser()
manualClean = [f.split(l) for l in manualClean]
num = dict()
num['s'] = sum(['s' in s for s in getSampleNames])
num['t'] = sum(['t' in s for s in getSampleNames])
regsets = dict()
chrs = set([l[1] for l in manualClean])
for samp in getSampleNames:
for chrom in chrs:
sub = [l for l in manualClean if l[7]==samp and l[1] == chrom]
regset = [[int(l[2]),int(l[3]), l[10], l[6]] for l in sub]
if 's' in samp:
if not ('s',chrom) in regsets.keys():
regsets['s', chrom] = list()
regsets['s',chrom].append(regset)
else:
if not ('t',chrom) in regsets.keys():
regsets['t', chrom] = list()
regsets['t',chrom].append(regset)
a = dict()
for chrom in chrs:
a['s',chrom]=intersRegs([sorted(s) for s in regsets['s',chrom]])
a['t',chrom]=intersRegs([sorted(s) for s in regsets['t',chrom]])
return a
reformat2
Dummy filter to skip manual clean phaseLast build was: Sun Jan 8 03:51:58 2012.
Required CPU time: 00:00:1.83.
def reformat2(ifile):
'Dummy filter to skip manual clean phase'
f, header = PennCNVParser()
ofile = list()
ofile.append('\t'.join(header))
p = getPairings()
for line in ifile:
newline = (f.split(line)[1:-1])
try:
t=newline.pop(10)
except IndexError:
print line
if newline[6] in [x[0] for x in p]:
newline.insert(7,'s')
else:
newline.insert(7,'t')
genes = newline[10].split(',')
for gene in genes:
newline[10]=gene
ofile.append('\t'.join(newline))
return ofile
genoTypeCheck
Builds a matrix of % of genotype differences between each couple of samplesLast build was: Sun Jan 8 19:42:16 2012.
Required CPU time: 00:02:57.52.
def genoTypeCheck(fnames):
"""Builds a matrix of % of genotype differences between each couple of samples"""
data=[]
for ifile in fnames:
curgtype = list()
cursamp = open(ifile).readlines()
for line in cursamp:
line = line.split('\t')
curgtype.append(line[3])
data.append(curgtype)
data=array(data)
differences=list()
f = open('gtypecheck_small.txt', 'w')
sdict = idToNameDict()
for sample1 in data:
templist = list()
for sample2 in data:
templist.append(float(sum(array(sample1)!=array(sample2))) / len(sample1) * 100)
try:
print sdict[sample1[0].replace('.GType','')], 'vs', sdict[sample2[0].replace('.GType','')],':','\t', templist[-1], '\t'
#print templist[-1]
if templist[-1] < 20 and sdict[sample1[0].replace('.GType','')] != sdict[sample2[0].replace('.GType','')]:
f.write(sdict[sample1[0].replace('.GType','')] + ' vs ' + sdict[sample2[0].replace('.GType','')] + '\t' + str(templist[-1]) + '\n')
differences.append(templist)
f = open('gtypecheck.txt', 'w')
for idx, item in enumerate(differences):
f.write(sdict[data[idx][0].replace('.GType','')]+' ')
f.write('\n')
for idx, item in enumerate(differences):
f.write(' ' + sdict[data[idx][0].replace('.GType','')])
f.writelines(str(array(item)).strip('[]').replace('\n','')+'\n')
except:
print 'Skipping couple: ' + sample1[0] +', '+sample2[0]
return differences
getGeneNames
Extract the names of all annotated genesLast build was: Sun Jan 8 03:47:16 2012.
Required CPU time: 00:00:0.05.
def getGeneNames(f):
"""Extract the names of all annotated genes"""
data=list()
for line in f:
nline=line[0:-1]
t = nline.split()
data.extend(t[7].split(','))
data=array(data)
return unique(data)
countCNVlenghts
Computes percentage of genome affected by CNVLast build was: Sun Jan 8 19:43:38 2012.
Required CPU time: 00:00:0.36.
def countCNVlenghts(getSampleNames, reformat):
"""Computes percentage of genome affected by CNV"""
di = idToNameDict()
y=[l.split('\t') for l in reformat]
glength=float(2867188341)
res = []
for s in getSampleNames:
try:
sub=[l for l in y if l[6]==s]
lens = [int(l[2])-int(l[1]) for l in sub]
res.append([di[s] , str(sum(lens)) , str(100*(sum(lens) / glength))])
except:
import pdb; pdb.set_trace()
for st in ['s', 't']:
sub = [l for l in y if l[7]==st]
lens = [int(l[2])-int(l[1]) for l in sub]
if st=='s':
label = 'Blood'
else:
label = 'Tumor'
print label + ' average: ' + str(sum(lens) /
(len(getSampleNames)/float(2))) +'\t' + str(100 * (sum(lens)/ (len(getSampleNames)/float(2)) / glength) )
return res
addStats
Adds descriptive statisticsLast build was: Sun Jan 8 03:47:17 2012.
Required CPU time: 00:00:0.99.
def addStats(fname):
"""Adds descriptive statistics"""
data=list()
f = fname
for line in f:
line=line[0:-1]
data.append(line.split('\t'))
data=array(data)
genes = unique(data[:,10])
p = getPairings()
s=list()
for samp in p[0]:
for raw in data:
if raw[6] == samp:
s.append(raw)
s=array(s)
t=list()
for samp in p[1]:
for raw in data:
if raw[6] == samp:
t.append(raw)
t=array(t)
gene_count = dict()
for gene in genes:
if gene == 'NOT_FOUND':
gene_count[gene]=(0,0,0,0)
else:
s_where = s[nonzero(s[:,10]==gene)]
t_where = t[nonzero(t[:,10]==gene)]
s_count = sum(s[:,10]==gene)
t_count = sum(t[:,10]==gene)
s_diffcount = len(unique(s_where[:,6]))
t_diffcount = len(unique(t_where[:,6]))
gene_count[gene]=(s_count, t_count, s_diffcount, t_diffcount)
of=list()
for idx, line in enumerate(data):
if idx==0:
of.append('\t'.join(line) + '\ts hits \tt hits\thits diff.\ts un. hits\tt un. hits\t un. hits diff.')
else:
currinfo = gene_count[line[10]]
of.append('\t'.join(line)+\
'\t'+str(currinfo[0])+\
'\t'+str(currinfo[1])+\
'\t'+str(abs(currinfo[0]-currinfo[1]))+\
'\t'+str(currinfo[2])+\
'\t'+str(currinfo[3])+\
'\t'+str(abs(currinfo[2]-currinfo[3])))
#'\t'+str(pvalue(currinfo[2], 8-currinfo[2], currinfo[3], 8-currinfo[3]).two_tail)+'\n')
return of
exportCNVlenghts
[documentation not available]Output file: cnv_lenghts.tsv
Last build was: Sun Jan 8 19:43:38 2012.
Required CPU time: 00:00:0.02.
def exportCNVlenghts(countCNVl):
f = open('cnv_lenghts.tsv', 'w')
for line in countCNVl:
f.writelines(str(line).strip('[]').replace(',','\t').replace("'",'')+'\n')
return 'cnv_lenghts.tsv'
exportDistMats
Exports the output of samplesDistMats to csv formatOutput files: inters_mat.txt , notIntersMat.txt , tanimotoMat.txt .
Last build was: Sun Jan 8 19:43:38 2012.
Required CPU time: 00:00:0.03.
def exportDistMats(samples, mats):
"""Exports the output of samplesDistMats to csv format"""
intMat=mats[0]
notIntMat=mats[1]
tanimotoMat=mats[2]
f_intMat = open('intersMat.txt','w')
f_notIntMat = open('notIntersMat.txt','w')
f_tanimotoMat = open('tanimotoMat.txt','w')
f_intMat.write(' ,'+','.join(samples) + '\n')
f_notIntMat.write(' ,'+','.join(samples) + '\n')
f_tanimotoMat.write(' ,'+','.join(samples) + '\n')
for idx, item in enumerate(intMat):
f_intMat.write(samples[idx]+',')
f_intMat.write(str(list(intMat[idx])).strip('[]').replace('.0','').replace(' ','')+'\n')
f_notIntMat.write(samples[idx]+',')
f_notIntMat.write(str(list(notIntMat[idx])).strip('[]').replace('.0','').replace(' ','')+'\n')
f_tanimotoMat.write(samples[idx]+',')
f_tanimotoMat.write(str(list(tanimotoMat[idx])).strip('[]').replace(' ','')+'\n')
return ['inters_mat.txt', 'notIntersMat.txt', 'tanimotoMat.txt']
intersMat_genes
Produces a list of common genes between each couple of samplesLast build was: Sun Jan 8 03:47:19 2012.
Required CPU time: 00:00:0.38.
def intersMat_genes(dataset, fname, genes, samples):
"""Produces a list of common genes between each couple of samples"""
inters_names = list()
for idx1, geneline1 in enumerate(dataset):
inters_names_t = list()
for idx2, geneline2 in enumerate(dataset):
inters_names_t.append(genes[nonzero(logical_and(geneline1, geneline2))])
inters_names.append(inters_names_t)
return inters_names
reformat
Change format from region-centric to gene-centricLast build was: Sun Jan 8 03:46:56 2012.
Required CPU time: 00:00:0.69.
def reformat(ifile):
"""Change format from region-centric to gene-centric"""
f, header = PennCNVParser()
ofile = list()
ofile.append('\t'.join(header))
p = getPairings()
for line in ifile:
newline = (f.split(line)[1:-1])
try:
t=newline.pop(10)
except IndexError:
print line
if newline[6] in [x[0] for x in p]:
newline.insert(7,'s')
else:
newline.insert(7,'t')
genes = newline[10].split(',')
for gene in genes:
newline[10]=gene
ofile.append('\t'.join(newline))
return ofile
CNVboolVects
Creates a matrix of presence/absence of CNV in each sample for each gene (out of all the gene that are affected by CNV in at least 1 sampleLast build was: Sun Jan 8 03:47:18 2012.
Required CPU time: 00:00:0.35.
def CNVboolVects(raw, allsamples):
"""Creates a matrix of presence/absence of CNV in each sample for each gene (out of all the gene that are affected by CNV in at least 1 sample"""
data=list()
f=raw
for line in f:
line=line[0:-1]
data.append(line.split('\t'))
data=array(data)
genes = unique(data[1:-1,10])
hashd = dict( zip( genes, range( 0,len(genes) ) ) )
#samples = getSampleNames()
samples = allsamples
s=data
vectors = np.zeros((len(samples),len(genes)))
for s_idx, sample in enumerate(samples):
sample_genes = s[s[:,6]==sample][:,10]
for gene in sample_genes:
vectors[s_idx][hashd[gene]]=1
return vectors
LogR_BAF_FileName
The name of the file containing Log R Ratio and B Allele Frequency dataOutput file: pglfeb.csv
Last build was: Sun Jan 8 03:37:36 2012.
Required CPU time: 00:00:0.03.
def LogR_BAF_FileName():
"""The name of the file containing Log R Ratio and B Allele Frequency data"""
return(fname)
clustergram
Calls R to make a clustergramOutput files: clustergram_001_108.png , clustergram_005_248.png , clustergram_05_1606.png , heatmap_001_108.png , heatmap_005_248.png , heatmap_05_1606.png .
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Last build was: Sun Jan 8 19:44:49 2012.
Required CPU time: 00:00:57.72.
def clustergram(diffmat, fisher, genes, samples):
"""Calls R to make a clustergram"""
#samplesNoLetter = [item.replace('t','').replace('s','') for item in samples]
#samples = []
#[samples.append(i) for i in samplesNoLetter if not i in samples]
p=getPairings()
sdict = idToNameDict()
s2 = list()
for i in range(0,len(p)):
s2.append(sdict[p[i][0]]+' vs '+sdict[p[i][1]])
samples = s2
d=diffmat[0]
gdict = geneChrDict()
#import pdb; pdb.set_trace()
thresholds = [0.01, 0.05, 0.5]
cfnames = list()
hfnames = list()
for t in thresholds:
ok_genes=list()
ok_gnames=list()
for idx, gene_row in enumerate(d):
if fisher[1][idx] < t:
if str.lower(genes[idx]) != 'loc100130872-spon2' and str.lower(genes[idx]) != 'loc647859':
ok_genes.append(gene_row)
ok_gnames.append(genes[idx])
print 'Selected genes: ' + str(len(ok_gnames))
for idx, gene in enumerate(ok_gnames):
if len(gdict[gene])==1:
chr = '0'+ gdict[gene]
else:
chr = gdict[gene]
ok_gnames[idx]= chr + ' ' + gene
ok_genes=array(ok_genes)
r.library('gplots')
colors = r.redgreen(len(unique(ok_genes)))
colors = colors[::-1]
#colors[2]='#880000'
colors[3]='#880000'
#return ok_genes, samples, ok_gnames, colors
cfnames.append('clustergram_' + str(t).replace('.','') + '_' + str(len(ok_gnames)) + '.png')
hfnames.append('heatmap_' + str(t).replace('.','') + '_' + str(len(ok_gnames)) + '.png')
callRforClustergram(cfnames[-1], ok_genes, samples, ok_gnames, colors)
makeHeatmap(hfnames[-1], ok_genes, samples, ok_gnames, colors)
return hstack([cfnames, hfnames])
PennCNV
Runs PennCNV to detect CNV regionsOutput files: pglfeb.csv_11s.tab.out , pglfeb.csv_11t.tab.out , pglfeb.csv_12s.tab.out , pglfeb.csv_12t.tab.out , pglfeb.csv_13s.tab.out , pglfeb.csv_13t.tab.out , pglfeb.csv_14Bt.tab.out , pglfeb.csv_14s.tab.out , pglfeb.csv_19s.tab.out , pglfeb.csv_19t.tab.out , pglfeb.csv_1s.tab.out , pglfeb.csv_1t.tab.out , pglfeb.csv_20s.tab.out , pglfeb.csv_20t.tab.out , pglfeb.csv_21s.tab.out , pglfeb.csv_21t.tab.out , pglfeb.csv_2s.tab.out , pglfeb.csv_2t.tab.out , pglfeb.csv_32s.tab.out , pglfeb.csv_32t.tab.out , pglfeb.csv_33s.tab.out , pglfeb.csv_33t1.tab.out , pglfeb.csv_34s.tab.out , pglfeb.csv_34t.tab.out , pglfeb.csv_36s.tab.out , pglfeb.csv_36t.tab.out , pglfeb.csv_37s.tab.out , pglfeb.csv_37t.tab.out , pglfeb.csv_3s.tab.out , pglfeb.csv_3t.tab.out , pglfeb.csv_43s.tab.out , pglfeb.csv_43t.tab.out , pglfeb.csv_44s.tab.out , pglfeb.csv_44t.tab.out , pglfeb.csv_45s.tab.out , pglfeb.csv_45t.tab.out , pglfeb.csv_4s.tab.out , pglfeb.csv_4t.tab.out , pglfeb.csv_5Bt.tab.out , pglfeb.csv_5s.tab.out , pglfeb.csv_5t.tab.out , pglfeb.csv_6s.tab.out , pglfeb.csv_6t.tab.out , pglfeb.csv_7s.tab.out , pglfeb.csv_7t.tab.out , pglfeb.csv_8s.tab.out , pglfeb.csv_8t.tab.out .
Last build was: Sun Jan 8 19:10:54 2012.
Required CPU time: 00:52:42.29.
def PennCNV(inputfiles):
"""Runs PennCNV to detect CNV regions"""
ofiles = list()
for ifile in inputfiles:
cmdstr = 'perl ' + penncnv_path + \
'/detect_cnv.pl -test -hmm ' + \
penncnv_path + '/lib/hhall.hmm -pfb ' + \
penncnv_path + '/lib/ho1v1.hg18.pfb -gcmodelfile ' + \
penncnv_path + '/lib/ho1v1.hg18.gcmodel ' + \
ifile + \
' -log "' + ifile + '.log" ' + \
'-out "' + fname + '_' + ifile + '.out"'
print 'Executing: ' + cmdstr
#print 'WARNING: skipping PennCNV call! Uncomment to really call.'
os.system(cmdstr)
#ofiles.append(lp.newFile(fname + '_' + ifile + '.out'))
ofiles.append(fname + '_' + ifile + '.out')
return ofiles
getSampleNames
Extracts sample namesLast build was: Sun Jan 8 03:47:10 2012.
Required CPU time: 00:00:0.03.
def getSampleNames(fname):
"""Extracts sample names"""
b=open(fname).readlines()
t=list()
p = getPairedSampleIDs()
for l in b[1:]:
s = l.split()[0]
#if s in p:
t.append(s)
return t
CompareRegionAndGenes
Computes intersections between regions and genesLast build was: Sun Jan 8 03:47:09 2012.
Required CPU time: 00:00:13.22.
def CompareRegionAndGenes(data):
"""Computes intersections between regions and genes"""
ref = open(penncnv_path+'/ucsc_hg18/refGene.txt').readlines()
for idx, line in enumerate(ref):
ref[idx] = line.split()
refgenes = [ref[i][12] for i in range(0, len(ref))]
res = list()
for idx, line in enumerate(data[1:]):
if mod(idx, 100)==0:
print 'Intersecting: '+str(idx)+' of '+str(len(data)-1)
line = line.split('\t')
gene = line[10]
try:
where = refgenes.index(gene)
int1 = regIntersect([int(line[1]), int(line[2])], [int(ref[where][4]), int(ref[where][5])])
int2 = regIntersect([int(line[1]), int(line[2])], [int(ref[where][6]), int(ref[where][7])])
#res.append(['***********************************************'])
res.append([
gene,
line[6], #sample
line[7], #s/t
int(line[1]), #reg start
int(line[2]), #reg end
int(line[2])-int(line[1]), # reg size
int(ref[where][4]), #trans start
int(ref[where][5]), #trans end
int(ref[where][5])-int(ref[where][4]),
int(ref[where][6]), #code start
int(ref[where][7]), #code end
int(ref[where][7])-int(ref[where][6]),
int1, #int trans
int2, #int code
])
except:
res.append([
gene,
line[6],
line[7],
int(line[1]),
int(line[2]),
'?', '?', '?', '?', '?', '?','?','?'
])
return res
exportFigExtRegs
Creates a figure for the extended regions using ROutput file: extRegs.pdf
Last build was: Sun Jan 8 19:45:02 2012.
Required CPU time: 00:00:3.74.
def exportFigExtRegs(frags):
"""Creates a figure for the extended regions using R"""
r.library('quantsmooth')
chrwidth = 10
chrspacing = 30
r.pdf(file='extRegs.pdf', height=10, width=12)
#maxarmlen = max(
maxlen = max([a[0][1] for a in frags['1']])
r.plot(1,type="n",xlim=[0,(chrspacing+chrwidth)*(len(frags)-1)],ylim=[0, maxlen],xaxt="n",yaxt="n", ann=False, frame_plot=False, tick_number=0, xlab="Autosomes")
keys = [int(k) for k in frags]
skeys = sort(keys)
tickat = list()
for cidx,chrom in enumerate([str(k) for k in skeys]):
offs = cidx*(chrspacing+chrwidth)
#maxy = max([a[0][1] for a in frags[chrom]])
tothits = sum([a[0][1]-a[0][0] for a in frags[chrom][0::2]])
maxlen = max([a[0][1] for a in frags[chrom]])
maxy = r.lengthChromosome(chrom, units = 'bases')[chrom]
r.lines([offs, offs], [0,maxy])
for fidx, frag in enumerate(frags[chrom]):
if frag[1]=='' and frag[2]=='':
continue
y = (frag[0][0]+float(frag[0][1]))/2
y = (y * maxy) / maxlen
l = frag [0][1]-frag[0][0]+1
lB = int(frag[1])
lT = int(frag[2])
Ts = sum([a=='T' for a in frag[3]])
Bs = sum([a=='B' for a in frag[3]])
#Ts_p = sum([a=='T' for idx,a in enumerate(frag[3]) if int(frag[4][idx])>2])
#Ts_n = sum([a=='T' for idx,a in enumerate(frag[3]) if int(frag[4][idx])<2])
#Bs_p = sum([a=='B' for idx,a in enumerate(frag[3]) if int(frag[4][idx])>2])
#Bs_n = sum([a=='B' for idx,a in enumerate(frag[3]) if int(frag[4][idx])<2])
Ts_p = sum([a=='T' for idx,a in enumerate(frag[3]) if int(frag[4][idx])>2])
Ts_n = sum([a=='T' for idx,a in enumerate(frag[3]) if int(frag[4][idx])<2])
Bs_p = sum([a=='B' for idx,a in enumerate(frag[3]) if int(frag[4][idx])>2])
Bs_n = sum([a=='B' for idx,a in enumerate(frag[3]) if int(frag[4][idx])<2])
Tgain = Ts_p - Ts_n
Bgain = Bs_p - Bs_n
#Tcol = 'red' if Tgain < 0 else 'green'
#Bcol = 'red' if Bgain < 0 else 'green'
Dcol = 'darkred' if Tgain - Bgain > 0 else 'darkgreen'
armlen = int(abs(lT - lB))*1.5
if max(lB, lT)>0:
if Dcol == 'darkred':
r.lines([offs +chrwidth-2 , armlen + offs +chrwidth-2], [maxy-y,maxy-y], col=Dcol)
else:
r.lines([offs -armlen -chrwidth+2, offs -chrwidth+2], [maxy-y,maxy-y], col=Dcol)
tickat.append(offs)
print chrom, tothits/float(maxy)
bleachv = 1-min((tothits/float(maxy))/0.08,1)
r.paintCytobands(chrom, pos=[offs+chrwidth/2, maxy], width=chrwidth, units="bases", orientation='v', legend=False, bleach=bleachv)
# r.axis(at=tickat, labels=range(1,23), cex_axis=1, side=range(1,23), mgp=[3,.5,0], xlab='Autosomes', main='Genomic regions affected by CNV')
r.axis(at=tickat, lty=0, labels=range(1,23), cex_axis=1, side=range(1,23), xlab='Autosomes', main='Genomic regions affected by CNV', mgp=[3,-0.8,0])
# if Bgain:
# #r.lines([-log10(l) + offs, 0 + offs], [y,y], col=Bcol)
# r.lines([offs - lB -1, 0 + offs -1], [y,y], col=Bcol)
# r.lines([offs - lB -1, 0 + offs -1], [y,y], col=Bcol)
# if Tgain:
# #r.lines([0 + offs, log10(l) + offs], [y,y], col=Tcol)
# r.lines([0 + offs +1 , lT + offs +1], [y,y], col=Tcol)
# r.lines([0 + offs +1 , lT + offs +1], [y,y], col=Tcol)
r.dev_off()
return('extRegs.pdf')
manualClean
Removes a list of CNVi-related genes produced elsewhereLast build was: Sun Jan 8 03:46:55 2012.
Required CPU time: 00:00:3.48.
def manualClean(x):
"""Removes a list of CNVi-related genes produced elsewhere"""
no=[int(n) for n in open('manuallyRemovedCNVs.txt').read().split()]
cleandata = list()
f = open('cleanedCNVs.txt', 'w')
for idx, line in enumerate(x):
if not (idx+1 in no):
cleandata.append(line)
f.write(line)
return cleandata
samplesDistMats
Computes Tanimoto distance between samplesLast build was: Sun Jan 8 03:47:21 2012.
Required CPU time: 00:00:0.24.
def samplesDistMats(dataset):
"""Computes Tanimoto distance between samples"""
inters_mat = squareform(pdist(dataset, lambda x,y: sum(logical_and(x,y))))
for idx in range(0,dataset.shape[0]):
inters_mat[idx,idx]=sum(dataset,axis=1)[idx]
notIntersMat = squareform(pdist(dataset, lambda x,y: sum(~logical_and(x,y))))
tanimotoMat = squareform(pdist(dataset, lambda x,y: 1 - sum(logical_and(x,y))/float(sum(logical_or(x,y))) ))
return [inters_mat, notIntersMat, tanimotoMat]
exportPerRegion_clean
Dummy node to skip manual filtering phaseOutput file: perRegion_clean.csv
Last build was: Sun Jan 8 19:45:02 2012.
Required CPU time: 00:00:0.14.
def exportPerRegion_clean(data):
"""Dummy node to skip manual filtering phase"""
f = open('perRegion_clean.csv','w')
sdict = idToNameDict()
f.write(data[0].replace('\t',',')+'\n')
for line in data[1:]:
l = line.replace(',','.').split()
l[6] = sdict[l[6]]
l = str(l).replace('\'', '').strip('[]')
f.write(l+'\n')
return 'perRegion_clean.csv'
exportCNVDiffMat
Exports to tsv fileOutput file: CNVgenes.csv
Last build was: Sun Jan 8 19:45:15 2012.
Required CPU time: 00:00:12.21.
def exportCNVDiffMat(cnvdiff, cnvdata, fisher, genes, samples):
"""Exports to tsv file"""
p = getPairings()
samples = list()
for a in p:
samples.extend([a[0], a[1]])
samples = samplesToNames(samples)
data = list()
for line in cnvdata:
data.append(line.split('\t'))
data = array(data)
vects, vs, vt = cnvdiff[0], cnvdiff[1], cnvdiff[2]
gdict = geneChrDict()
for i in range(len(samples),0,-1):
if mod(i,2)==0:
samples.insert(i, samples[i-1]+'-'+samples[i-2])
header =['gene', 'Chr',
'B gain', 'B loss', 'B CNV',
'T gain', 'T loss', 'T CNV',
'B CNV + T CNV',
'TvsB loss','TvsB gain', 'TvsBgain+TvsBloss', 'TvsB loss/gain p-value',
'max CNV', 'Reg. Max length',
'CNV samples (repeated if hit in multiple regions)']
header = hstack([header, samples])
fisher_abs = fisher[0]
fisher_diff = fisher[1]
f = open('CNVgenes.csv', 'w')
f.write(str(header).strip('[]').replace('\n','').replace('\' \'',',').strip('\'') + '\n')
for idx, vect in enumerate(vects):
#gene name
f.write(genes[idx])
#if genes[idx]=='ACOT11':
# import pdb; pdb.set_trace()
#chromosome
f.write(',' + str(gdict[genes[idx]]))
#cnv stats
cnvBgain = sum(vs[idx]>2)
cnvBloss = sum(vs[idx]<2)
cnvB = cnvBgain + cnvBloss
cnvTgain = sum(vt[idx]>2)
cnvTloss = sum(vt[idx]<2)
cnvT = cnvTgain + cnvTloss
f.write(', ' + str(cnvBgain) + ',' + str(cnvBloss) + ',' + str(cnvB))
f.write(', ' + str(cnvTgain) + ',' + str(cnvTloss) + ',' + str(cnvT))
f.write(', ' + str(cnvB+cnvT))
# f.write(', ' + str(cnvB) + ',' + str(cnvT) + ',' + str(cnvB+cnvT))
#fisher on B vs T cnvs
#f.write(','+str(fisher_abs[idx]))
#differential cnvs stats
f.write(str(',' +
str(sum(vect<0))+','+
str(sum(vect>0))+','+
str(sum(vect<0)+
sum(vect>0))+',')
.replace('\n',''))
#fisher on s vs t differential cnvs
f.write(',' + str(fisher_diff[idx]) + ',')
# max CNV
cns = hstack([vs[idx],vt[idx]])
maxcnv = cns[argmax(abs(2-cns))]
f.write(str(maxcnv) + ',')
# per gene additional info
info = extractPerGeneProperties(data, genes[idx])[1:]
for idx2, item in enumerate(info):
if not item=='nan':
if idx2 in [3,4,5]:
f.write(item.replace(',','').replace('\n','').replace('\'', '').replace('[','').replace(']','')+',')
#cnv per sample
f.write(str(zip(vs[idx],vt[idx],vect)).strip('[]').replace('(','').replace(')','').replace('\n','').replace('.0',''))
f.write('\n')
return 'CNVgenes.csv'
exportMergedFragments
Exports extended regions to tsv fileOutput file: extRegions.tsv
Last build was: Sun Jan 8 19:45:15 2012.
Required CPU time: 00:00:0.09.
def exportMergedFragments(mf):
'''Exports extended regions to tsv file'''
f = open('extRegions.tsv', 'w')
f.write('Chr.\t'+
'ExtReg. Start\t'+
'ExtReg. End\t'+
'ExtReg. Length\t'+
'Max hits B\t'+
'Max hits T\t'+
'B/T\t'+
'CN\t'+
'"Genes"\n')
for chrom in mf:
for extreg in mf[chrom]:
f.write(
#chromosome
chrom+'\t'+
#reg start
str(extreg[0][0])+'\t'+
#reg end
str(extreg[0][1])+'\t'+
#reg length
str(extreg[0][1]-extreg[0][0]+1)+'\t'+
# max hits B
str(extreg[1])+'\t'+
# max hits T
str(extreg[2])+'\t'+
#BT
str(extreg[3]).strip('[]').replace("'",'').replace(',','')+'\t'+
#CN
str(extreg[4]).strip('[]').replace("'",'').replace(',','')+'\t'+
#genes
str(extreg[5]).replace('set','').replace('(','').replace(')','')
.replace('[','').replace(']','').replace("'",'')+'\n'
)
return 'extRegions.tsv'
exportMergedRegsTB
Exports to tsv fileOutput file: mergedRegsTB.tsv
Last build was: Sun Jan 8 19:45:16 2012.
Required CPU time: 00:00:0.66.
def exportMergedRegsTB(mergedRegs):
"""Exports to tsv file"""
f = open('mergedRegs.tsv', 'w')
f.write('Chr.\tReg. Start\tReg. End\tReg. Lenght\tNum Samples\tCN-B\tCN-T\tnB\tnT\t"Genes"\n')
for key in sorted(mergedRegs.keys()):
for i in range(0, len(mergedRegs[key][0])):
# try:
try:
cnb = [cn for idx, cn in enumerate(mergedRegs[key][3][i])
if mergedRegs[key][4][i][idx]=='B']
cnt = [cn for idx, cn in enumerate(mergedRegs[key][3][i])
if mergedRegs[key][4][i][idx]=='T']
except:
import pdb; pdb.set_trace()
f.write(
# Chr
key+'\t'+
#Reg. Start e Reg. End
str(mergedRegs[key][0][i]).strip('[]').replace(', ','\t')+'\t'+
#Reg. Length
str(mergedRegs[key][0][i][1]-mergedRegs[key][0][i][0]+1)+'\t'+
#Num Samples
str(mergedRegs[key][1][i])+'\t'+
#CN-B
str(cnb).
replace('[','').
replace(']','').
replace("'", '')+'\t'+
#CN-T
str(cnt).
replace('[','').
replace(']','').
replace("'", '')+'\t'+
#nB
str(sum([st=='B' for st in mergedRegs[key][4][i]]))+'\t'+
#nT
str(sum([st=='T' for st in mergedRegs[key][4][i]]))+'\t'+
#Genes
str(mergedRegs[key][2][i]).replace('set','').
replace(')','').
replace('(','').
replace('[','').
replace(']','').
replace("'", '')+'\n'
)
# except: import pdb; pdb.set_trace()
return 'mergedRegsTB.tsv'
mergeFragments
Merges all contiguous non-void region fragmentsLast build was: Sun Jan 8 19:44:58 2012.
Required CPU time: 00:00:0.37.
def mergeFragments(intRegs):
"""Merges all contiguous non-void region fragments"""
extreg=dict()
for chrom in intRegs:
extreg[chrom] = list()
currExtRegBT = list()
currExtRegCN = list()
currExtRegS = [Inf,0]
currExtRegG = set()
currExtRegMaxhitsB = 0
currExtRegMaxhitsT = 0
for idx, frag in enumerate(intRegs[chrom][4]):
a = intRegs[chrom][0][idx][0]
b = intRegs[chrom][0][idx][1]
#import pdb; pdb.set_trace()
if frag != []:
currExtRegMaxhitsT = max(currExtRegMaxhitsT, sum([a1=='T' for a1 in frag]))
currExtRegMaxhitsB = max(currExtRegMaxhitsB, sum([a1=='B' for a1 in frag]))
currExtRegBT.extend(frag)
currExtRegCN.extend(intRegs[chrom][3][idx])
currExtRegS = [min(currExtRegS[0], a), max(currExtRegS[1],b)]
currExtRegG = currExtRegG.union(intRegs[chrom][2][idx])
else:
extreg[chrom].append([currExtRegS, currExtRegMaxhitsB,
currExtRegMaxhitsT, currExtRegBT, currExtRegCN, currExtRegG])
extreg[chrom].append([[a,b], '', '', '', '', 'unknown'])
currExtRegCN=list()
currExtRegBT=list()
currExtRegG = set()
currExtRegS = [Inf,0]
currExtRegMaxhitsT = 0
currExtRegMaxhitsB = 0
# handle last fragment (will get no '[]' as stop signal)
if frag != []:
extreg[chrom].append([currExtRegS, currExtRegMaxhitsB,
currExtRegMaxhitsT, currExtRegBT, currExtRegCN, currExtRegG])
return extreg
computeFisher
Tests the hyphotesis that a gene is hit by CNV in M samples out of N by chanceLast build was: Sun Jan 8 19:43:52 2012.
Required CPU time: 00:00:8.96.
def computeFisher(cnvect):
"""Tests the hyphotesis that a gene is hit by CNV in M samples out of N by chance"""
svec = cnvect[1]
tvec = cnvect[2]
diff = cnvect[0]
numsamp = len(svec[0])
fishval_diff = zeros(svec.shape[0])
fishval_abs = zeros(svec.shape[0])
for idx, row in enumerate(diff):
a11 = sum(row < 0)
a12 = numsamp-a11
a21 = sum(row > 0)
a22 = numsamp-a21
#fishval[idx]=r.fisher_test(array([[a11,a12],[a21,a22]]), alternative='less')['p.value']
fishval_diff[idx]=r.fisher_test(array([[a11,a12],[a21,a22]]))['p.value']
for idx, row in enumerate(svec):
a11 = sum(row != 2)
a12 = numsamp-a11
a21 = sum(tvec[idx] != 2)
a22 = numsamp-a21
fishval_abs[idx]=r.fisher_test(array([[a11,a12],[a21,a22]]))['p.value']
return fishval_abs, fishval_diff
prepareInput
Converts data to a format suitable for PennCNVOutput files: 11s.tab , 11t.tab , 12s.tab , 12t.tab , 13s.tab , 13t.tab , 14Bt.tab , 14s.tab , 19s.tab , 19t.tab , 1s.tab , 1t.tab , 20s.tab , 20t.tab , 21s.tab , 21t.tab , 2s.tab , 2t.tab , 32s.tab , 32t.tab , 33s.tab , 33t1.tab , 34s.tab , 34t.tab , 36s.tab , 36t.tab , 37s.tab , 37t.tab , 3s.tab , 3t.tab , 43s.tab , 43t.tab , 44s.tab , 44t.tab , 45s.tab , 45t.tab , 4s.tab , 4t.tab , 5Bt.tab , 5s.tab , 5t.tab , 6s.tab , 6t.tab , 7s.tab , 7t.tab , 8s.tab , 8t.tab .
Last build was: Sun Jan 8 03:42:33 2012.
Required CPU time: 00:04:57.23.
def prepareInput(fname, ssheet):
"""Converts data to a format suitable for PennCNV"""
ss = [l.split('\t') for l in open(ssheet).readlines()][1:]
scolumns = range(4,200,3)
#a=open(fname).readline()
#samples = [line.replace('.GType','') for line in a.split(',')[3::3]]
samples = [s[0] for s in ss]
fnames = list()
for k, sample in enumerate(samples):
print 'Preparazione campione ' + sample + '...',
#print 'SKIPPING!'
os.system('cut -f 1-3,' + str(scolumns[k]) + '-' + str(scolumns[k]+2) + ' --delimiter , "' + fname +'" > ' + sample + '.csv')
os.system('cat ' + sample + ".csv | tr ',' '\t' > " + sample + '.tab')
os.system('rm ' + sample + '.csv')
fnames.append(sample + '.tab')
return fnames
exportFigCNVlenghts
Creates a figure of the samples based % of genome affected by CNVOutput files: cnvlenghts.pdf , cnvlenghts_log.pdf .
Last build was: Sun Jan 8 19:45:16 2012.
Required CPU time: 00:00:0.04.
def exportFigCNVlenghts(countCNVlenghts_o):
"""Creates a figure of the samples based % of genome affected by CNV"""
r.pdf('cnvlenghts_log.pdf')
y = [int(x[1])/1000000 for x in countCNVlenghts_o if not 'B' in x[0]]
l = [x[0] for x in countCNVlenghts_o if not 'B' in x[0]]
yidx = sorted(range(len(y)), key = y.__getitem__)
ys = sorted(y)
ls = [l[x] for x in yidx]
r.barplot(ys, horiz=True, names_arg=ls, log='x', cex_names=0.5, las=1)
r.dev_off()
r.pdf('cnvlenghts.pdf')
y = [int(x[1]) for x in countCNVlenghts_o if not 'B' in x[0]]
l = [x[0] for x in countCNVlenghts_o if not 'B' in x[0]]
yidx = sorted(range(len(y)), key = y.__getitem__)
ys = sorted(y)
ls = [l[x] for x in yidx]
r.barplot(ys, horiz=True, names_arg=ls, cex_names=0.5, las=1)
#r.lines(range(0,len(ys)), r.cumsum(ys))
#r.lines(r.cumsum(y), range(0,len(y)))
r.dev_off()
return ('cnvlenghts.pdf', 'cnvlenghts_log.pdf')
makeBed
Create Bed file for use with Genome BrowserOutput file: pglfeb.csv.bed
Last build was: Sun Jan 8 19:45:16 2012.
Required CPU time: 00:00:0.42.
def makeBed(CNVinput):
"""Create Bed file for use with Genome Browser"""
os.system('perl ' + penncnv_path + '/visualize_cnv.pl -format bed -output "' + fname + '.bed" "' + CNVinput + '"')
return fname + '.bed'
CNVDiffMat
Makes a matrix of B/T difference in CN between same genes of different samplesLast build was: Sun Jan 8 19:43:41 2012.
Required CPU time: 00:00:2.43.
def CNVDiffMat(fname, genes, allsamples):
"""Makes a matrix of B/T difference in CN between same genes of different samples"""
data=list()
f=fname
for line in f:
line=line[0:-1]
data.append(line.split('\t'))
data=array(data)
hashd = dict( zip( genes, range(0,len(genes)) ) )
p = getPairings()
sp = [x[0] for x in p]
tp = [x[1] for x in p]
s = list()
for sample in sp:
for line in data:
if sample == line[6]:
s.append(line)
t = list()
for sample in tp:
for line in data:
if sample == line[6]:
t.append(line)
samples = sp
s_vectors = np.zeros((len(samples),len(genes)))*nan
s_vectors_min =np.zeros((len(samples),len(genes)))*nan
s_vectors_num =np.zeros((len(samples),len(genes)))*nan
s_vectors_cn = np.zeros((len(samples),len(genes)))*nan
for s_idx, sample in enumerate(samples):
print s_idx, 'of', len(samples),',',
x = nonzero([l[6]==sample for l in s])[0]
sub_s = [s[pickme] for pickme in x]
sub_s = array(sub_s)
sample_genes = sub_s[:,10]
for gene in unique(sample_genes):
if isnan(s_vectors[s_idx][hashd[gene]]):
temp = array(sub_s[sub_s[:,10]==gene][:,5], int)
maxidx = argmax(temp)
minidx = argmin(temp)
absmax = temp[maxidx]
absmin = temp[minidx]
s_vectors[s_idx][hashd[gene]] = absmax
s_vectors_min[s_idx][hashd[gene]] = absmin
s_vectors_cn[s_idx][hashd[gene]] = assignGeneCN(temp)
s_vectors_num[s_idx][hashd[gene]] = len(temp)
s_vectors[isnan(s_vectors)] = 2
s_vectors_min[isnan(s_vectors_min)] = 2
s_vectors_cn[isnan(s_vectors_cn)] = 2
s_vectors_num[isnan(s_vectors_num)] = 0
samples = tp
t_vectors = np.zeros((len(samples),len(genes)))*nan
t_vectors_min = np.zeros((len(samples),len(genes)))*nan
t_vectors_num =np.zeros((len(samples),len(genes)))*nan
t_vectors_cn = np.zeros((len(samples),len(genes)))*nan
for t_idx, sample in enumerate(samples):
print t_idx, 'of', len(samples),',',
x = nonzero([l[6]==sample for l in t])[0]
sub_t = [t[pickme] for pickme in x]
sub_t = array(sub_t)
try:
sample_genes = sub_t[:,10]
except IndexError:
import pdb; pdb.set_trace()
for gene in unique(sample_genes):
#if gene == 'PC':
# import pdb; pdb.set_trace()
if isnan(t_vectors[t_idx][hashd[gene]]):
temp = array(sub_t[sub_t[:,10]==gene][:,5], int)
maxidx = argmax(temp)
absmax = temp[maxidx]
minidx = argmin(temp)
absmin = temp[minidx]
t_vectors[t_idx][hashd[gene]] = absmax
t_vectors_min[t_idx][hashd[gene]] = absmin
t_vectors_num[t_idx][hashd[gene]] = len(temp)
t_vectors_cn[t_idx][hashd[gene]] = assignGeneCN(temp)
t_vectors[isnan(t_vectors)]=2
t_vectors_min[isnan(t_vectors_min)] = 2
t_vectors_cn[isnan(t_vectors_cn)] = 2
t_vectors_num[isnan(t_vectors_num)] = 0
return [t_vectors_cn.T-s_vectors_cn.T, #0
s_vectors_cn.T, t_vectors_cn.T, #1,2
s_vectors_min.T, t_vectors_min.T, #3,4
s_vectors_num.T, t_vectors_num.T, #5,6
s_vectors.T, t_vectors.T] #7,8