Parsing¶
Bioinformatics, and parsing and storing collections of (flat) files... we are worth better than that.
Yet another FASTQ, FASTA, <name it> set of parsers¶
Yes, this is just an other parser. The twist here is the attempt to change the way the data is approached by establishing the following guiding principles:
- Do not care about the file format, but about the information to be extracted
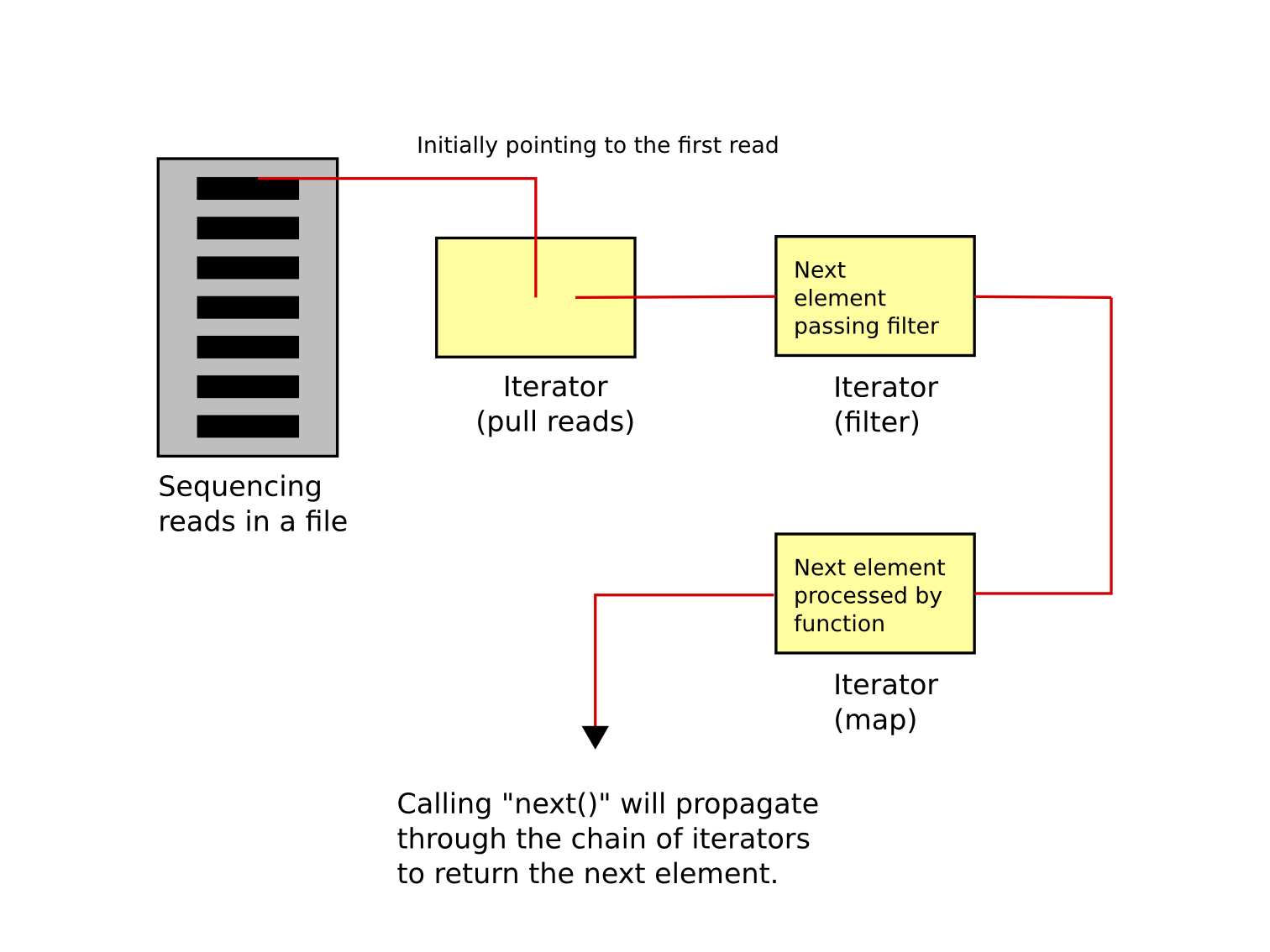
- Forget the sacrosanct atomic step (flat) file -> tool -> (flat) file and consider the chaining of data streams (of objects). Evaluation is lazy. What you need will be fetched when you need it.
The first principle is implemented in the function parsing.open():
# We want to parse out data
from ngs_plumbing import parsing
filename = 'foo.fastq'
# Open the file, guessing the file format from the extension
sequence_data = parsing.open(filename)
Note
filename could have been a FASTA file, compressed with gzip or bzip2, or a SAM/BAM file (although this is a little less tested at the moment), and it would have worked with the same exact call:
filename = 'foo.fastq.gz'
sequence_data = parsing.open(filename)
The resulting object sequence_data can be iterated through, and data extracted.
For example, if we want the sequence and its quality:
# Iterator through sequences+quality
seq_stream = sequence_data.iter_seqqual()
The iterator will return objects with attributes sequence and quality.
No matter how big the sequencing file is, the memory usage remains low. This is leading to the second principle: chaining and lazy evaluation with iterators.
The next entry in the iterator can be called with next():
entry = next(seq_stream)
# length for the read
len(entry.sequence)
Note
Performances are reasonable for my needs: a FASTQ file of 0.430 Gb can be iterated through in 4 seconds on my development laptop (iCore 7, Python 3.3). That’s slower than the empirical theoretical limit, which is the counting of lines in the file with
wc -l
This is running on 0.13s (after repeated access, so with the file buffered by the system). Using a benchmark on Biostar, we calculate that this is only 6.7 times slower than the fastest C implementation (seqtk), which is not bad (~100 Mb / second) considering that this is pure Python (with all the benefits of being usable from Python). The low-level interface in biopython (Bio.SeqIO.QualityIO import FastqGeneralIterator) is achieving similar performances as ours on that benchmark (I could not get the current biopython to install on Python 3.3, and so could not verify it).
If speed is a bottleneck, get in touch.
Pulling entries into a pipeline¶
A common operation is to filter reads according to quality. We can just implement a filter (return True is passing the the filter, False otherwise):
AVG_QUAL = 200
def has_avgqual(x):
# bytes. Summing them is summing their ASCII codes
quality = x.quality
return (sum(quality) / len(quality)) > AVG_QUAL
filter(has_avgqual, seq_stream)
Filtering on read lengths (useful on Ion Torrent data, for example) is similar:
LENGTH = 150
def is_long(x):
# bytes. Summing them is summing their ASCII codes
return len(x.sequence) > LENGTH
filter(has_avgqual, is_long)
An other possible operation is to look for barcode ‘ATACAGAC’, located 33 bases after the begining of the read (yes, molecular biology tricks make such, or even weirder, situations occur).
def has_barcode(entry):
return entry.sequence.startswith('ATACAGAC', 33)
Now we can write the combined filter as:
barcoded_stream = filter(has_barcode,
filter(has_avgqual, seq_stream))
The evaluation is lazy, and query the first good barcoded read will only unwind the stream of sequences (and check the average quality and the presence of a barcode) until the first such read is found. No intermediate flat files, the ability to quickly check interactively if anything appears to be coming out of it:
good_read = next(barcoded_stream)
Note
When performances matter, combining simple filters into one function rather than chain the filter might make a difference.
Now let’s imagine for a second that an alignment algorithm for short reads is/can be accessed as a Python module (we shall call it groovyaligner). This module could have a function align() that takes Python objects with attributes sequence and quality:
import groovyaligner as ga
mapping_stream = map(ga.align, barcoded_stream)
Now mapping_stream would be an iterator of alignments. Again, no flat file involved, no parsing, and actually no computation done until the program starts pulling elements one by one through the chain of iterators.
Note
There is actually a “groovyaligner” in the work to be included in this package.
This would let one inspect the final outcome of chained iterators without computing completely all intermediate steps, something we believe to be really valuable when working interactively:
from itertools import islice
# takes the result of the 10 first mappings
islice(mapping_stream, 0, 10)
Paired-end data¶
Paired-end data are often stored in two files. The iterators can work just fine.
# We want to parse out data
from ngs_plumbing import parsing
filename_1 = 'foo_s_1_1.fastq'
filename_2 = 'foo_s_1_2.fastq'
# Open the file, guessing the file format from the extension
sequence_pairs = zip(parsing.open(filename_1),
parsing.open(filename_2))
Warning
With Python 2, zip() is not returning a generator but will get _all_ pairs. Use the itertools.izip().
Note
Depending on the sample preparation, one might want to change the orientation of one of the reads in the pair.
- def revcomp(x):
- # reverse complement of x.sequence
Getting the next pair is then:
pair = next(sequence_pairs)
Formats¶
The handling of file formats is restricted to NGS data, and and oriented toward what we consider the most common operations with such data.
For a complete set of parsers for practically all existing formats in bioinformatics, there is biopython.