Welcome to the OBITools¶
The OBITools package is a set of programs specifically designed for analyzing
NGS data in a DNA metabarcoding context, taking into account taxonomic
information. It is distributed as an open source software available on the
following website: http://metabarcoding.org/obitools.
Citation: Boyer F., Mercier C., Bonin A., Taberlet P., Coissac E. (2014) OBITools: a Unix-inspired software package for DNA metabarcoding. Molecular Ecology Resources, submitted.
Installing the OBITools¶
Availability of the OBITools¶
The OBITools are open source and protected by the CeCILL 2.1 license
(http://www.cecill.info/licences/Licence_CeCILL_V2.1-en.html).
The OBITools are deposited on the Python Package Index (PyPI : https://pypi.python.org/pypi/obitools)
and all the sources can be downloaded from our subversion server
(http://www.grenoble.prabi.fr/public-svn/OBISofts/OBITools).
Prerequisites¶
To install the OBITools, you need that these softwares are installed on your
system:
- Python 2.7 (installed by default on most
Unixsystems, available from the Python website) gcc(installed by default on mostUnixsystems, available from the GNU sites dedicated to GCC and GMake)
On a linux system¶
You have to take care that the Python-dev packages are installed.
On MacOSX¶
The C compiler and all the other compilation tools are included in the XCode
application not installed by default. The Python included in the system is not
suitable for running the OBITools. You have to install a complete distribution
of Python that you can download as a MacOSX package from the Python website.
Downloading and installing the OBITools¶
The OBITools are downloaded and installed using the get-obitools.py script.
This is a user level installation that does not need administrator privilege.
Once downloaded, move the file get-obitools.py in the directory where you want to install
the OBITools. From a Unix terminal you must now run the command :
> python get-obitools.py
The script will create a new directory at the place you are running it in which all the
OBITools will be installed. No system privilege are required, and you system will not
be altered in any way by the obitools installation.
The newly created directory is named OBITools-VERSION where version is substituted by the latest version number available.
Inside the newly created directory all the OBITools are installed. Close to this directory
there is a shell script named obitools. Running this script activate the OBITools
by reconfiguring your Unix environment.
> ./obitools
Once activated you can desactivate the OBITools byt typing the command exit.
> exit OBITools are no more activated, Bye... ======================================
System level installation¶
To install the OBITools at the system level you can follow two options :
- copy the
obitoolsscript in a usual directory for installing program like/usr/local/binbut never move theOBIToolsdirectory itself after the installation by theget-obitools.py.- The other solution is to add the
export/bindirectory located in theOBIToolsdirectory to the ``PATH``environment variable.
Retrieving the sources of the OBITools¶
If you want to compile by yourself the OBITools, you will need to install the same
prerequisite:
> pip install -U virtualenv > pip install -U sphinx > pip install -U cython
moreover you need to install any subversion client (a list of clients is available from Wikipedia)
Then you can download the
> svn co http://www.grenoble.prabi.fr/public-svn/OBISofts/OBITools/branches/OBITools-1.00/ OBITools
This command will create a new directory called OBITools.
Introduction¶
DNA metabarcoding is an emerging approach for biodiversity studies (Taberlet et
al. 2012). Originally mainly developed by microbiologists (e.g. Sogin et al.
2006), it is now widely used for plants (e.g. Sonstebo et al. 2010, Parducci et
al. 2012, Yoccoz et al. 2012) and animals from meiofauna (e.g. Chariton et al.
2010, Baldwin et al. 2013) to larger organisms (e.g. Andersen et al. 2012,
Thomsen et al. 2012). Interestingly, this method is not limited to sensu
stricto biodiversity surveys, but it can also be implemented in other
ecological contexts such as for herbivore (e.g. Valentini et al. 2009, Kowalczyk
et al. 2011) or carnivore (e.g. Deagle et al. 2009, Shehzad et al. 2012) diet
analyses.
Whatever the biological question under consideration, the DNA metabarcoding
methodology relies heavily on next-generation sequencing (NGS), and generates
considerable numbers of DNA sequence reads (typically million of reads).
Manipulation of such large datasets requires dedicated programs usually running
on a Unix system. Unix is an operating system, whose first version was created
during the sixties. Since its early stages, it is dedicated to scientific
computing and includes a large set of simple tools to efficiently process text
files. Most of those programs can be viewed as filters extracting information
from a text file to create a new text file. These programs process text files as
streams, line per line, therefore allowing computation on a huge dataset without
requiring a large memory. Unix programs usually print their results to their
standard output (stdout), which by default is the terminal, so the results can
be examined on screen. The main philosophy of the Unix environment is to allow
easy redirection of the stdout either to a file, for saving the results, or to
the standard input (stdin) of a second program thus allowing to easily create
complex processing from simple base commands. Access to Unix computers is
increasingly easier for scientists nowadays. Indeed, the Linux operating system,
an open source version of Unix, can be freely installed on every PC machine and
the MacOSX operating system, running on Apple computers, is also a Unix system.
The OBITools programs imitate Unix standard programs because they usually act as
filters, reading their data from text files or the stdin and writing their
results to the stdout. The main difference with classical Unix programs is that
text files are not analyzed line per line but sequence record per sequence
record (see below for a detailed description of a sequence record).
Compared to packages for similar purposes like mothur (Schloss et al. 2009) or
QIIME (Caporaso et al. 2010), the OBITools mainly rely on filtering and sorting
algorithms. This allows users to set up versatile data analysis pipelines
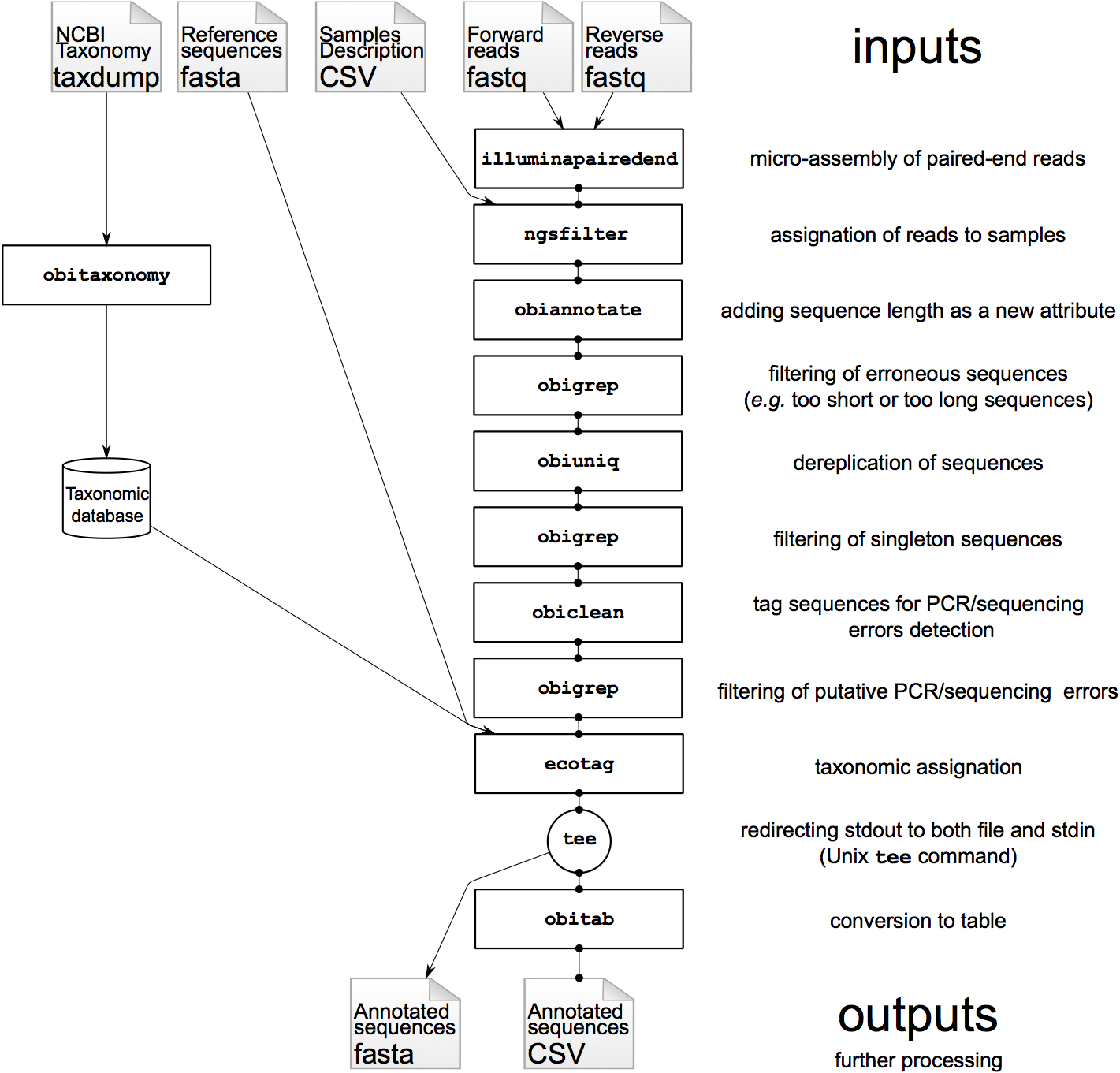
(Figure 1), adjustable to the broad range of DNA metabarcoding applications.
The innovation of the OBITools is their ability to take into account the
taxonomic annotations, ultimately allowing sorting and filtering of sequence
records based on the taxonomy.
References¶
Andersen K, Bird KL, Rasmussen M, Haile J, Breuning-Madsen H, Kj�r KH, Orlando L, Gilbert MTP, Willerslev E (2012) Meta-barcoding of “dirt” DNA from soil reflects vertebrate biodiversity. Molecular Ecology, 21, 1966-1979.
Baldwin DS, Colloff MJ, Rees GN, Chariton AA, Watson GO, Court LN, Hartley DM, Morgan Mj, King AJ, Wilson JS, Hodda M, Hardy CM (2013) Impacts of inundation and drought on eukaryote biodiversity in semi-arid floodplain soils. Molecular Ecology, 22, 1746-1758.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Tumbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7, 335-336.
Chariton AA, Court LN, Hartley DM, Colloff MJ, Hardy CM (2010) Ecological assessment of estuarine sediments by pyrosequencing eukaryotic ribosomal DNA. Frontiers in Ecology and the Environment, 8, 233-238.
Deagle BE, Kirkwood R, Jarman SN (2009) Analysis of Australian fur seal diet by pyrosequencing prey DNA in faeces. Molecular Ecology, 18, 2022-2038.
Kowalczyk R, Taberlet P, Coissac E, Valentini A, Miquel C, Kaminski T, W�jcik JM (2011) Influence of management practices on large herbivore diet - case of European bison in Bialowieza Primeval Forest (Poland). Forest Ecology and Management, 261, 821-828.
Parducci L, Jorgensen T, Tollefsrud MM, Elverland E, Alm T, Fontana SL, Bennett KD, Haile J, Matetovici I, Suyama Y, Edwards ME, Andersen K, Rasmussen M, Boessenkool S, Coissac E, Brochmann C, Taberlet P, Houmark-Nielsen M, Larsen NK, Orlando L, Gilbert MTP, Kjaer KH, Alsos IG, Willerslev E (2012) Glacial Survival of Boreal Trees in Northern Scandinavia. Science, 335, 1083-1086.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75, 7537-7541.
Shehzad W, Riaz T, Nawaz MA, Miquel C, Poillot C, Shah SA, Pompanon F, Coissac E, Taberlet P (2012) Carnivore diet analysis based on next generation sequencing: application to the leopard cat (Prionailurus bengalensis) in Pakistan. Molecular Ecology, 21, 1951-1965.
Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta JM, Herndl GJ (2006) Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proceedings of the National Academy of Sciences of the United States of America, 103, 12115-12120.
S�nsteb� JH, Gielly L, Brysting A, Reidar E, Edwards M, Haile J, Willerslev E, Coissac E, Rioux D, Sannier J, Taberlet P, Brochmann C (2010) Using next-generation sequencing for molecular reconstruction of past Arctic vegetation and climate. Molecular Ecology Resources, 10, 1009-1018.
Taberlet P, Coissac E, Hajibabaei M, Rieseberg LH (2012) Environmental DNA. Molecular Ecology, 21, 1789-1793.
Thomsen PF, Kielgast J, Iversen LL, Wiuf C, Rasmussen M, Gilbert MTP, Orlando L, Willerslev E (2012) Monitoring endangered freshwater biodiversity using environmental DNA. Molecular Ecology, 21, 2565-2573.
Valentini A, Miquel C, Nawaz MA, Bellemain E, Coissac E, Pompanon F, Gielly L, Cruaud C, Nascetti G, Wincker P, Swenson JE, Taberlet P (2009) New perspectives in diet analysis based on DNA barcoding and parallel pyrosequencing: the trnL approach. Molecular Ecology Resources, 9, 51-60.
Yoccoz NG, Br�then KA, Gielly L, Haile J, Edwards ME, Goslar T, von Stedingk H, Brysting AK, Coissac E, Pompanon F, S�nsteb� JH, Miquel C, Valentini A, de Bello F, Chave J, Thuiller W, Wincker P, Cruaud C, Gavory F, Rasmussen M, Gilbert MTP, Orlando L, Brochmann C, Willerslev E, Taberlet P (2012) DNA from soil mirrors plant taxonomic and growth form diversity. Molecular Ecology, 21, 3647-3655.
Basic concepts of the OBITools¶
Once installed, the OBITools enrich the Unix command line interface with a set
of new commands dedicated to NGS data processing. Most of them have a name
starting with the obi prefix. They automatically recognize the input file
format amongst most of the standard sequence file formats (i.e. fasta, fastq,
EMBL, and GenBank formats). Nevertheless, options are available to enforce some
format specificity such as the encoding system used in fastq files for quality
codes. Most of the basic Unix commands have their OBITools equivalent (e.g.
obihead vs head, obitail vs tail, obigrep vs grep), which is
convenient for scientists familiar with Unix. The main difference between any
standard Unix command and its OBITools counterpart is that the treatment unit is
no longer the text line but the sequence record. As a sequence record is more
complex than a single text line, the OBITools programs have many supplementary
options compared to their Unix equivalents.
The structure of a sequence record¶
The OBITools commands consider a sequence record as an entity composed of five
distinct elements. Two of them are mandatory, the identifier (id) and the DNA or
protein sequence itself. The id is a single word composed of characters, digits,
and other symbols like dots or underscores excluding spaces. Formally, the ids
should be unique within a dataset and should identify each sequence record
unambiguously, but only a few OBITools actually rely on this property. The
sequence is an ordered set of characters corresponding to nucleotides or
amino-acids according to the International Union of Pure and Applied Chemistry
(IUPAC) nomenclature (Cornish-Bowden 1985). The three other elements composing a
sequence record are optional. They consist in a sequence definition, a quality
vector, and a set of attributes. The sequence definition is a free text
describing the sequence briefly. The quality vector associates a quality score
to each nucleotide or amino-acid. Usually this quality score is the result of
the base-calling process by the sequencer. The last element is a set of
attributes qualifying the sequence, each attribute being described by a
key=value pair. The set of attributes is the central concept of the OBITools
system. When an OBITools command is run on the sequence records included in a
dataset, the result of the computation often consist in the addition of new
attributes completing the annotation of each sequence record. This strategy of
sequence annotation allows the OBITools to return their results as a new
sequence record file that can be used as the input of another OBITools program,
ultimately creating complex pipelines.
Managed sequence file formats¶
Most of the OBITools commands read sequence records from a file or from the
stdin, make some computations on the sequence records and output annotated
sequence records. As inputs, the OBITools are able to automatically recognize
the most common sequence file formats (i.e. fasta, fastq, EMBL, and GenBank).
They are also able to read ecoPCR (Ficetola et al. 2010) result files and
ecoPCR/ecoPrimers formatted sequence databases (Riaz et al. 2011) as
ordinary sequence files. File format outputs are more limited. By default,
sequences without and with quality information are written in fasta and Sanger
fastq formats, respectively. However, dedicated options allow enforcing the
output format, and the OBITools are also able to write sequences in the
ecoPCR/ecoPrimers database format, to produce reference databases for these
programs. In the fasta or fastq format, the attributes are written in the header
line just after the id, following a key=value; format (Figure 2).

Taxonomical aspects¶
Filtering and annotation steps in the processing of DNA metabarcoding sequence
data are greatly eased by the explicit association of taxonomic information to
sequences together with an easy access to the taxonomy. Taxonomic information,
including a taxonomic identifier, can thus be stored in the set of attributes of
each sequence record. Specifically, the taxid attribute is used by the
OBITools when querying taxonomic information of a sequence record, nevertheless
several OBITools commands can annotate sequence records with taxonomy-related
attributes for the user’s convenience. The value of the taxid attribute must
be a unique integer referring unambiguously to one taxon in the taxonomic
associated database. Although this is not mandatory, the NCBI taxonomy is a
preferred source of taxonomic information as the OBITools provide commands to
easily extract the full taxonomic information from it. The command obitaxonomy
is useful to build a taxonomic database in the OBITools format from a dump of
the NCBI taxonomic database (downloadable at the following URL:
ftp://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz).
Implemented algorithms¶
Most of the algorithms implemented in the OBITools are basic algorithms allowing
sampling, filtering and annotation of sequence records based on their associated
attribute set or sequence (e.g. obisample, obigrep, obiannotate). Some
others implement algorithms directly related to NGS or to DNA metabarcoding
(e.g. illuminapairedend, ngsfilter, ecotag). Finally, a few of them do not
run on sequence records and/or do not provide their results as sequence records.
Amongst them, oligotag (Coissac 2012) generates a set of short oligonucleotide
sequences (hereafter referred to as tags useful to uniquely identify
individual samples within a single NGS library containing many samples. Hereby,
we will describe some of the implemented algorithms pertaining directly to DNA
metabarcoding, as well as the corresponding programs. A full description of all
programs included in the OBITools suite is available on the web
http://metabarcoding.org/obitools/doc.
Implementation of the OBITools¶
The OBITools are a set of Python programs relying on an eponym Python library.
The OBITools library is mainly developed in Python (version 2.7 see
(http://www.python.org). For increasing the speed of execution, many parts of
the OBITools library are developed using cython (http://cython.org/, a Python
to C compiler) or the C language directly. The OBITools compile on Unix systems
including Linux and MacOSX.
References¶
Coissac E (2012) Oligotag: a program for designing sets of tags for next-generation sequencing of multiplexed samples. In: Data Production and Analysis in Population Genomics: Methods and Protocols (eds. Pompanon F, Bonin A), pp. 13-31. Springer Science+Business Media, New York.
Cornish-Bowden A (1985) Nomenclature for incompletely specified bases in nucleic acid sequences: recommendations 1984. Nucleic Acids Research, 13, 3021-3030.
Ficetola GF, Coissac E, Zundel S, Riaz T, Shehzad W, Bessi�re J, Taberlet P, Pompanon F (2010) An in silico approach for the evaluation of DNA barcodes. BMC Genomics, 11, 434.
Riaz T, Shehzad W, Viari A, Pompanon F, Taberlet P, Coissac E (2011) ecoPrimers: inference of new DNA barcode markers from whole genome sequence analysis. Nucleic Acids Research, 39, e145.