6. phaseshifts API¶
6.1. Package Contents¶
This chapter covers the main modules of the phaseshifts and provides some API documentation for those wishing to incorporate this package into their own projects.
6.2. Subpackages¶
- The main sub packages are listed below:
- phaseshifts.gui - includes all the necessary files for the graphical user interface.
- phaseshifts.lib - contains the Fortran libphsh library and the python wrappings.
- phaseshifts.doc - source documentation for the phaseshifts package.
- phaseshifts.test - modules for testing the phaseshift package.
6.3. Submodules¶
6.3.1. phaseshifts.atorb¶
atorb.py
Provides convenience functions for generating input and calculating atomic charge densities for use with the Barbieri/Van Hove phase shift calculation package.
| See: | http://www.icts.hkbu.edu.hk/surfstructinfo/SurfStrucInfo_files/leed/ |
|---|---|
| Requires: | f2py (for libphsh fortran wrapper generation) |
Note
To generate libphsh fortran wrappers (libphsh.pyd) for your platform then use ‘python setup.py’ in the lib directory of this package to install into your python distribution. Alternatively, use:
f2py -c -m libphsh libphsh.f
Windows users may have to add appropriate compiler switches, e.g.
f2py -c -m libphsh --fcompiler=gfortran --compiler=mingw-32 libphsh.f
- class phaseshifts.atorb.Atorb(**kwargs)[source]¶
Bases: object
Notes
Original author: Eric Shirley
There are nr grid points, and distances are in Bohr radii
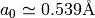
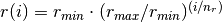 ,
,
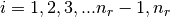
The orbitals are stored in phe(), first index goes
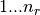 , the
second index is the orbital index (
, the
second index is the orbital index (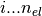 )
)Look at the atomic files after printing this out to see everything... Suffice it to say, that the charge density at radius
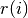 in units of electrons per cubic Bohr radius is given by:
in units of electrons per cubic Bohr radius is given by: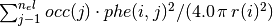
Think of the phe functions as plotting the radial wave-functions as a function of radius on a logarithmic mesh...
The Dirac equation is solved for the orbitals, whereas their density is treated by setting
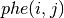 to Dirac’s
to Dirac’s
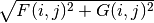 times the sign of
times the sign of 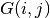 ...
...So we are doing Dirac-Fock, except that we are not treating exchange exactly, in terms of working with major and minor components of the orbitals, and the phe’s give the CORRECT CHARGE DENSITY...
The above approximation ought to be very small for valence states, so you need not worry about it...
The Breit interaction has been neglected altogether...it should not have a huge effect on the charge density you are concerned with...
- static calculate_Q_density(**kwargs)[source]¶
Parameters: kwargs may be any of the following. :
element : int or str, optional
Generate element atorb input file on the fly. Additional kwargs may be used to govern the structure of the input file - please use help(phaseshifts.Atorb.gen_input) for more information.
input : str, optional
Specify atorb input file otherwise will use the class instance value.
output_dir : str, optional
Specify the output directory for the at_*.i file generated, otherwise the default current working directory is used.
Returns: str : filename
Examples
>>> Atorb.calculate_Q_density(input='atorb_C.txt') 18.008635 -33.678535 4.451786 -36.654271 1.569616 -37.283660 0.424129 -37.355634 0.116221 -37.359816 0.047172 -37.360317 0.021939 -37.360435 0.010555 -37.360464 0.005112 -37.360471 0.002486 -37.360473 0.001213 -37.360473 0.000593 -37.360473 0.000290 -37.360474 N L M J S OCC. 1 0 0 -1/2 1 2.0000 -11.493862 2 0 0 -1/2 1 2.0000 -0.788618 2 1 1 -1/2 1 0.6667 -0.133536 2 1 1 -3/2 1 1.3333 -0.133311 TOTAL ENERGY = -37.360474 -1016.638262
>>> Atorb.calculate_Q_density(element='H') 0.500007 -0.343752 0.152392 -0.354939 0.065889 -0.357254 0.028751 -0.357644 0.012732 -0.357703 0.005743 -0.357711 0.002641 -0.357712 0.001236 -0.357713 0.000587 -0.357713 0.000282 -0.357713 N L M J S OCC. 1 0 0 -1/2 1 1.0000 -0.229756 TOTAL ENERGY = -0.357713 -9.733932
- static gen_input(element, **kwargs)[source]¶
Parameters: element : int or str
Either the atomic number, symbol or name for a given element
output : str, optional
File string for atomic orbital output (default: ‘at_<symbol>.i’)
ngrid : int, optional
Number of points in radial grid (default: 1000)
rel : bool, optional
Specify whether to consider relativistic effects
filename : str, optional
Name for generated input file (default: ‘atorb’)
header : str, optional
Comment at beginning of input file
method : str, optional
Exchange correlation method using either 0.0=Hartree-Fock, 1.0=LDA, -alpha = xalpha (default: 0.0)
relic : float, optional
Relic value for calculation (default: 0)
mixing_SCF : float, optional
Self consisting field value (default: 0.5)
tolerance : float, optional
Eigenvalue tolerance (default: 0.0005)
ech : float, optional
(default: 100)
- static get_quantum_info(shell)[source]¶
Returns: tuple : (int, int, list[float, float], list[float, float])
(n, l, j=[l-s, l+s], occ=[
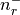 ,
, 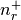 ])
])Notes
- n is the principle quantum number (
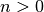 ).
). - l is the azimuthal quantum number (
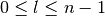 ).
). - s is the spin quantum number (
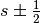 ).
). - j is the total angular momentum quantum numbers for both
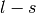 or
or 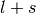 , respectively.
, respectively. 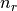 is the occupancy of the spin-split
and levels, respectively.
is the occupancy of the spin-split
and levels, respectively.
- n is the principle quantum number (
- static replace_core_config(electron_config)[source]¶
Parameters: electron_config : str
String containing the electronic configuration of the given element.
Returns: str : :
A substituted string where the nobel gas core has been replaced.
Examples
>>> Atorb.replace_core_config('[Ar] 4s2') '1s2 2s2 2p6 3s2 3p6 4s2'
>>> Atorb.replace_core_config('[Xe] 6s2 5d1') '1s2 2s2 2p6 3s2 3p6 3d10 4s2 4p6 5s2 4d10 5p6 6s2 5d1'
6.3.2. phaseshifts.conphas¶
conphas.py
Provides a native python version of the conphas (phsh3) FORTRAN program by W. Moritz, which is distributed as part of the SATLEED code (see “Barbieri/Van Hove phase shift calculation package” section) and can be found at: http://www.icts.hkbu.edu.hk/surfstructinfo/SurfStrucInfo_files/ leed/leedpack.html
The Conphas() class also provides a number of convenience functions (see docstrings below).
6.3.2.1. Examples¶
>>> from os.path import join
>>> from phaseshifts.conphas import Conphas
>>> con = Conphas(output_file=join('testing', 'leedph_py.d'),
lmax=10)
>>> con.set_input_files([join('testing', 'ph1')])
>>> con.set_format('cleed')
>>> con.calculate()
- class phaseshifts.conphas.Conphas(input_files=[], output_file=[], formatting=None, lmax=10, **kwargs)[source]¶
Class Conphas
Notes
This work is based on the original conphas (phsh3) FORTRAN program by W. Moritz, which is distributed as part of the SATLEED code (see “Barbieri/Van Hove phase shift calculation package” section) and can be found at: http://www.icts.hkbu.edu.hk/surfstructinfo/SurfStrucInfo_files/ leed/leedpack.html
- _Conphas__fix_path(file_path)¶
Fix escaped characters in filepath
- _Conphas__set_data(data=None)¶
- calculate()[source]¶
Calculates continuous phase shifts from input file(s).
Examples
>>> con = Conphas(output_file=r'testing\leedph_py.d', lmax=10) >>> con.set_input_files([r'testing\ph1']) >>> con.set_format('cleed') >>> con.calculate() L = 0 jump between 25.0 eV and 30.0 eV; IFAK = -1 L = 1 jump between 65.0 eV and 70.0 eV; IFAK = -1 L = 2 jump between 20.0 eV and 25.0 eV; IFAK = 1 jump between 80.0 eV and 85.0 eV; IFAK = 0 L = 3 L = 4 jump between 275.0 eV and 280.0 eV; IFAK = 1 L = 5 L = 6 L = 7 L = 8 L = 9 L = 10
- load_data(filename)[source]¶
Load (discontinuous) phase shift data from file
Parameters: file : str
Path to phase shift file.
Returns: tuple: (double, double, int, int, ndarray) :
(initial_energy, energy_step, n_phases, lmf, data)
Notes
- initial_energy is the starting energy of the phase shifts.
- energy_step is the change in energy between consecutive values.
- n_phases is the number of phase shifts contained in the file.
- lmf is the maximum azimuthal quantum number considered.
- data is a (2 x n_phases) array containing the phase shift data.
- read_datafile(filename)[source]¶
Read in discontinuous phase shift file
Parameters: filename : str
The path to the discontinuous phase shift file
- set_format(formatting=None)[source]¶
Set appropriate format from available options
Parameters: format : str, optional
The format identifier for different packages; can be ‘cleed’ or None.
6.3.3. phaseshifts.elements¶
Properties of the chemical elements.
Each chemical element is represented as an object instance. Physicochemical and descriptive properties of the elements are stored as instance attributes.
| Author: | Christoph Gohlke |
|---|---|
| Version: | 2013.03.18 |
6.3.3.1. Requirements¶
6.3.3.2. References¶
6.3.3.3. Examples¶
>>> from elements import ELEMENTS
>>> len(ELEMENTS)
109
>>> str(ELEMENTS[109])
'Meitnerium'
>>> ele = ELEMENTS['C']
>>> ele.number, ele.symbol, ele.name, ele.eleconfig
(6, 'C', 'Carbon', '[He] 2s2 2p2')
>>> ele.eleconfig_dict
{(1, 's'): 2, (2, 'p'): 2, (2, 's'): 2}
>>> sum(ele.mass for ele in ELEMENTS)
14659.1115599
>>> for ele in ELEMENTS:
... ele.validate()
... ele = eval(repr(ele))
6.3.4. phaseshifts.leed¶
Provides CLEED validator and Converter classes.
The CLEED_validator() class provides a method for checking the input files for errors, whereas the Converter.import_CLEED() method allows importing CLEED input files as a MTZ_model class
- class phaseshifts.leed.CLEED_validator[source]¶
Bases: object
Class for validation of CLEED input files
- class phaseshifts.leed.Converter[source]¶
Bases: object
Convert different input into phaseshift compatible input
- static import_CLEED(filename)[source]¶
Imports CLEED input file and converts model to muffin-tin input.
- It assumes the following:
the basis vectors a1, a2, & a3 are x,y,z cartezian coordinates
if no a3 is found, the maximum z distance between atoms multiplied by four is given
the unitcell is converted from cartezian to fractional coordinates
atom sites are converted from Angstrom to Bohr units
additional info from the phase shift filename is provided by splitting the ‘_’ chars:
- First string segment is element or symbol, e.g. Ni
- Second string segment is the oxidation, e.g. +2
lines with ‘rm:‘ provide the radii dictionary of the atomic species
if no ‘rm:‘ found for that species, the atomic radius is used for zero valence, otherwise the covalent radius is used.
Additional information can, however, be provided using ‘phs:’ at the start of a line within the input file and may have the following formats:
- “phs: c <float> nh <int> nform <int> exchange <float>“
- “phs: <phase_shift> valence <float> radius <float>“
The identifiers exchange, nform, valence and radius may be abbreviated to exc, nf, val and rad, respectively.
Parameters: filename : str
Path to input file.
Returns: phaseshifts.model.MTZ_model :
Raises: IOError : filename invalid
ValueError : bad formatting of input
6.3.5. phaseshifts.model¶
model.py
Provides convenience functions for generating input and calculating atomic charge densities for use with the Barbieri/Van Hove phase shift calculation package.
- class phaseshifts.model.Atom(element, coordinates=[0.0, 0.0, 0.0], **kwargs)[source]¶
Bases: object
Atom class for input into cluster model for muffin-tin potential calculations.
- exception phaseshifts.model.CoordinatesError(msg)[source]¶
Bases: exceptions.Exception
Coordinate exception to raise and log duplicate coordinates.
- class phaseshifts.model.MTZ_model(unitcell, atoms, **kwargs)[source]¶
Bases: phaseshifts.model.Model
Muffin-tin potential Model subclass for producing input file for muffin-tin calculations in the Barbieri/Van Hove phase shift calculation package.
- calculate_MTZ(mtz_string='', **kwargs)[source]¶
Parameters: atomic_file : str
The path to the atomic input file. If this is omitted the default is generate one using the MTZ_model.gen_atomic() method.
cluster_file : str
The path to the cluster input file which may be a bulk or slab model.
slab : int or bool
Determines whether the MTZ calculation is for a slab model (True). The default is a bulk calculation.
output : dict
Dictionary output of ‘mtz’ - muffin-tin potential & ‘output_file’ - the path to the MTZ output file.
Returns: output_files : list(str)
Paths to the MTZ output file.
- create_atorbs(**kwargs)[source]¶
Returns: output_files : dict
Dictionary list of atorb*.i input files for the Atorb class to calculate the charge density from.
- gen_atomic(**kwargs)[source]¶
Parameters: input_dir : str
Input directory where at*.i files are kept.
input_files : tuple
List of input files to generate atomic input file from.
output_file : str
The filename of the resulting atomic*.i output file, which is simply a superimposed set of the radial charge densities from the individual input files.
Returns: output_file : str
Returns the output file path string.
Raises: IOError : exception
If either input directory or files do not exist.
Notes
If ‘input_files’ is not given then the default list of input files are inferred from the list of atoms in the model.
- gen_input(**kwargs)[source]¶
Returns: filename on success :
Raises: CoordinatesError : exception
if the model atoms have duplicate coordinates and the ‘pos_check’ kwarg is set to True.
- load_from_file(filename)[source]¶
Parameters: filename : str
The path of the input file (e.g. cluster*.i or slab.i)
Raises: IOError : exception
If the file cannot be read.
TypeError : exception
If a input line cannot be parsed correctly.
- class phaseshifts.model.Model(unitcell, atoms, **kwargs)[source]¶
Bases: object
Generic model class.
- _nineq_atoms()[source]¶
Returns: nineq_atoms, element_dict : tuple
- nineq_atoms : The estimated number of inequivalent atoms based on
the valence and radius of each atom.
- element_dict : a dictionary of each element in the atom list where
each element contains an atom dictionary of ‘nineq_atoms’, ‘n_atoms’ and a complete ‘atom_list’
- add_atom(element, position, **kwargs)[source]¶
Append an Atom instance to the model
Parameters: element : str or int
Either an element name, symbol or atomic number.
position : list(float) or ndarray
(1x3) array of the fractional coordinates of the atom within the unit cell in terms of the lattice vector a.
- check_coordinates()[source]¶
Check for duplicate coordinates of different atoms in model.
Raises: CoordinateError : exception
If duplicate positions found.
- class phaseshifts.model.Unitcell(a, c, matrix_3x3, **kwargs)[source]¶
Bases: object
Unitcell class
- set_a(a)[source]¶
Parameters: a: float :
The magnitude of the in-plane lattice vector in Angstroms
Notes
To retrieve a in terms of Angstroms use ‘unitcell.a’, whereas the internal parameter ‘unitcell._a’ converts a into Bohr radii (1 Bohr = 0.529Å), which is used for the muffin-tin potential calculations in libphsh (CAVPOT subroutine).
- set_c(c)[source]¶
Parameters: c : float
The magnitude of the in-plane lattice vector in Angstroms
Notes
To retrieve c in terms of Angstroms use ‘unitcell.c’, whereas the internal parameter ‘unitcell._c’ converts c into Bohr radii (1 Bohr = 0.529Å), which is used for the muffin-tin potential calculations in libphsh (CAVPOT subroutine).